

Genetic variation of the hemagglutinin of avian influenza virus H9N2
Abstract
Avian influenza virus H9N2 has become the dominant subtype of influenza which is endemic in poultry. The hemagglutinin, one of eight protein-coding genes, plays an important role during the early stage of infection. The adaptive evolution and the positively selected sites of the HA (the glycoprotein molecule) of H9N2 subtype viruses were investigated. Investigating 68 hemagglutinin H9N2 avian influenza virus isolates in China and phylogenetic analysis, it was necessary that these isolates were distributed geographically from 1994, and were all derived from the Eurasian lineage. H9N2 avian influenza virus isolates from domestic poultry in China were distinct phylogenetically from those isolated in Hong Kong, including viruses which had infected humans. Seven amino acid substitutions (2T, 3T, 14T, 165D, 197A, 233Q, 380R) were identified in the HA possibly due to positive selection pressure. Apart from the 380R site, the other positively selected sites detected were all located near the receptor-binding site of the HA1 strain. Based on epidemiological and phylogenetics analysis, the H9N2 epidemic in China was divided into three groups: the 1994–1997 group, the 1998–1999 group, and the 2000–2007 group. By investigating these three groups using the maximum likelihood estimation method, there were more positive selective sites in the 1994–1997 and 1998–1999 epidemic group than the 2000–2007 groups. This indicates that those detected selected sites are changed during different epidemic periods and the evolution of H9N2 is currently slow. The antigenic determinant or other key functional amino acid sites should be of concern because their adjacent sites have been under positive selection pressure. The results provide further evidence that the pathogenic changes in the H9N2 subtype are due mainly to re-assortment with other highly pathogenic avian influenza viruses. J. Med. Virol. 83:838–846, 2011. © 2011 Wiley-Liss, Inc.
Abbreviations:
AIV, avian influenza virus; HPAI, highly pathogenic avian influenza; LPAI, low pathogenic avian influenza; HA, hemagglutinin; RBS, receptor-binding site; LRT, likelihood ratio test; PAML, phylogenetic analysis by maximum likelihood; AH, anhui; BJ, Beijing; JS, Jiangsu; SJZ, Shijiazhuang; NX, Ningxia; HN, Henan; HB, Hebei; SH, Shanghai; YN, Yunnan; XZ, Xuzhou; ZJ, Zhejiang; GS, Gansu; NJ, Nanjing; SD, Shandong; HLJ, Heilongjiang; TJ, Tianjing; GD, Guangdong; SZ, Shenzhen; HK, Hongkong; GX, Guangxi; SC, Sichuan; LN, Liaoning; SG, Shaoguan; ST, Shantou; GZ, Guangzhou; CK, chicken; DK, duck; SW, swine; GF, guineafowl; PG, pigeon; BD, bird.
INTRODUCTION
Avian influenza virus (AIV) was first described as early as 1900 and is a highly infectious virus among Avian species. AIV is an RNA virus which contains eight segmented single strand RNA elements with negative polarity. If poultry in a hennery is infected with AIV, all poultry in the same hennery will quickly be infected with the same AIV. This can increase mortality and decrease production. Because of the rapid mutation of AIV, a new virus strain would often be found in avian species along with many antigenic variants. Currently there is no effective vaccine to protect poultry against AIV. Furthermore, if the AIV and human influenza viruses can infect the same host, a new influenza virus strain could be produced by reassortment, and a human pandemic may result. Studies on the evolution of AIV based on the nucleotide or amino acid sequence may find a solution to prevent the possibility of an avian influenza epidemic [Chen et al., 2003; Choi et al., 2004; Li et al., 2005]. Among all subtypes of AIV, only H5 and H7 are highly pathogenic avian influenza (HPAI) viruses that can infect humans. The next potential candidate of the HPAI virus, H9N2, has circulated in a large area of the world and has received much attention [Chen et al., 2003].
Avian influenza virus H9N2 first identified in China in 1994 [Cheng et al., 2001; Matrosovich et al., 2001; Liu et al., 2002a], has become the principal AIV subtype of the epidemic among poultry in China. Although it is a low pathogenic avian influenza (LPAI) virus to date, H9N2 can circulate widely, and has the ability to co-infect poultry with other microorganisms. The evolution of H9N2 and its reassortment with another subtype of virus or with a human influenza virus, indicates that LPAI H9N2 may become an HPAI virus [Chen et al., 2003]. The H9N2 pandemic in poultry can decrease production of poultry and even increase mortality in poultry. The two human cases infected with H9N2 in Hong Kong in 1999 suggested that the pandemic potential of H9N2 influenza viruses in humans should have raised greater concerns [Saito et al., 2002].
The surface glycoprotein HA is the most important target for the avian immune system. The HA protein of AIV is composed of 560 amino acids, and is responsible for viral attachment and entry into host cells. Virulence requires that the HA cleaves into two subunits when infecting the host cells. The amino acid patterns in the cleavage site of the HA are different between the HPAI and LPAI [Cheng et al., 2001; Plotkin et al., 2002; Saito et al., 2002; Liu et al., 2002a,b].
Therefore, gradual mutations of the HA gene produce continuously immunologically distinct strains of the virus that cause annual outbreaks. Influenza infection can lead to lasting immunity to the infecting strain, but most poultry are susceptible to re-infection by a new strain within a few years [Plotkin et al., 2002; Saito et al., 2002]. The HA protein may be a major target for selective pressure because it is one of the best characterized proteins of AIV, which plays a key role in overcoming the species barrier and leading to interspecies transmission from animals to humans [Plotkin et al., 2002; Saito et al., 2002; Mann et al., 2007; Chen and Chen, 2008].
The hemagglutinin is an important glycoprotein found on the surface of the virus, because the amino acid residue site mutations in the protein domain can lead to the alterations of biological function were investigated.
The HA protein is an important antigenic determinant, consequently the adaptive evolution and positive selection of sites in the HA protein of H9N2 in China.
The large amount of sequence data regarding H9N2 available in the NCBI has provided us with an opportunity to investigate the properties and evolution of H9N2. A concentration of research investigating the genetic evolution and characteristics of H9N2 has been conducted [Guo et al., 2000; Lin et al., 2000; Plotkin et al., 2002; Choi et al., 2004; Alexander, 2007; Cong et al., 2007; Wu et al., 2008]. The type and strengths of selective force on different amino acid sites are different. However, the HA protein adaptive evolutionary processes of H9N2 at the molecular level in China and the exact positively selected sites associated with this process remain unknown.
RESULTS
Phylogenetic Tree
Two phylogenetic trees with a total 68 HA gene sequences were constructed in order by Seqboot.exe, Distance.exe, and Neighbor.exe from the PHYLIP software package [Felsenstein, 1989]. The replicates used in Seqboot are 100. One of the phylogenetic trees (Fig. 1), is the 48 HA gene sequences of AIV isolates between 1994 and 1999, when the H9N2 AIV only circulated in poultry and wild birds. The results showed that the early AIV was dispersed in China, and there were fewer relationships between these AIV isolates from 1994 to 1999. The other phylogenetic tree (Fig. 2) involved 68 HA genes in AIV isolates between 1994 and 2007 indicated that geographically, there is no significant clue that AIV had circulated in China as a result of trade transport.

Phylogenetic tree of AIV HA gene in China 1994–1999. Phylogenetic tree was constructed in ordinal by Seqboot.exe, Distance.exe, and Neighbor.exe from the PHYLIP software. This figure of the tree is displayed by the TreeView.

Phylogenetic tree of AIV HA gene in China 1994–2007. Phylogenetic tree was constructed in ordinal by Seqboot.exe, Distance.exe, and Neighbor.exe from the PHYLIP software. This figure of the tree is displayed by the TreeView.
Through phylogenetic analysis, HA genes in two human-infected isolates (H9N2 and AIV) in Hong Kong in 1999 (HK/1073/99 and HK/1074/99) were differentiated from the evolutionary trend in China. There is also direct evidence that the virus H9N2 infecting humans after 1999 were related to the evolution of the HA gene in the H9N2 subtype. The two HA genes in H9N2 and AIV from the human infections in Hong Kong might be rearranged with highly pathogenic AIV subtype co-infecting the same host cell.
During the last 13 years, the H9N2 subtype has undergone more rapid evolution than previously [Alexander, 2007; Mann et al., 2007]. In order to obtain the evolutionary information and characteristics of the HA protein in the H9N2 subtype, the evolutionary process of the HA protein in the epidemic H9N2 virus in poultry and mammals in China was classified into three epidemic periods: the 1994–1997 epidemic group; the 1998–1999 interspecies group; the 2000–2007 epidemic group. By investigating the three different H9N2 epidemic groups, information regarding the evolution of H9N2 in China from different periods can be obtained.
Detection of Recombination
Because intragenic recombination can influence the detection of positive selection greatly, intragenic recombination detection was implemented for the epidemic H9N2 (China, 1994–2007) dataset using RDP3.0 software [Martin and Rybicki, 2000]. For the 68 HA sequences, only the Ck/YN/nh/01 was detected as a recombinant of Dk/NJ/01/99 and Ck/GX/9/99. It is also significant that intragenic recombination was found in the HA gene of the H9N2 virus which had occurred previously. This finding should be validated using epidemiological data. This suspected recombinant isolate was excluded from further detection of positive selection in order to guarantee the success of analysis.
Detection of Selected Sites
The likelihood values, selective sites and parameter estimates of 67 HA protein sequences from six models, such as “one-ratio” implemented in the program codeml in the PAML3.15 Software Package [Yang, 1997], are listed in Table I. The average ω values range from 0.22 to 0.27 among all models, thus showing the evidence of purifying selection. The “one-ratio” model (M0) showed that all sites of HA protein have a ω ratio of 0.22, and it is rejected as a result of the lowest likelihood value (−9,288.41). Three models (M2a, M3, and M8) showed that positive selection is detected in the HA protein which contains 0.8–1.3% positively selected sites with similar ω values (4.37–6.16). In total, seven positive selection sites were detected in M3 and M8 models including 2T, 3T, 14T, 165D, 197A, 233Q, and 380R, and five positive selection sites were detected in M2a model including 2T, 3T, 197T, 233Q, and 380R.
| Model | LnL | dN/dS | Estimated parameters | 2Δl | Positive selected sites |
|---|---|---|---|---|---|
| M0(one-ratio) | −9,288.41 | 0.22 | ω = 0.22 | 451.66 | None |
| M3(discrete) | −9,062.58 | 0.23 | p0 = 0.67902, p = 0.30803, p2 = 0.01295 | (13.28) | 2T,3T,14T,165D,197A,233Q,380R |
| ω0 = 0.03716, ω1 = 0.49587, ω2 = 4.36616 | |||||
| M1a(NearlyNeural) | −9,118.66 | 0.22 | p0 = 0.85444, p1 = 0.14556 | 85.76 | None |
| M2a(PositiveSelection) | −9,075.78 | 0.27 | p0 = 0.84718, p1 = 0.14469, p2 = 0.00812 | (9.21) | 2T,3T,197A,233Q,380R |
| ω2 = 0.08903, ω2 = 1.00000, ω2 = 6.16378 | |||||
| M7(beta) | −9,116.04 | 0.22 | p = 0.21462, q = 0.76912 | 107.54 | None |
| M8(beta and ω) | −9,062.27 | 0.24 | p0 = 0.98833, p1 = 0.01167 | (9.21) | 2T,3T,14T,165D,197A,233Q,380R |
| p = 0.30402, q = 1.30556, ω = 4.59687 |
- The posterior probability of black boldface site is over 99%, the other is over 95%. The values in parentheses represent the significant level of 0.01 with a χ2 distribution at d.f. = 4 (M0 vs. M3) or 2 (M1a vs. M2a and M7 vs. M8). M0,M1a,M2a,M3,M7,M8 are site models which allow the ω ratio to vary among sites respectively. Above results were obtained by codeml from PAML software package version 3.15.
In order to validate the significance of the model, the likelihood comparison of the selected model and the neutral model (M0 vs. M3, M1 vs. M2a, M7 vs. M8) was conducted. The likelihood ratio test (LRT) statistic 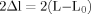 is employed to validate the significance of the results. LRT statistical results showed that the three selection models fit the data significantly better than the neutral models without selection.
is employed to validate the significance of the results. LRT statistical results showed that the three selection models fit the data significantly better than the neutral models without selection.
Posterior probability of each positive selection site (2T, 3T, 14T, 165D, 197A, 233Q and 380R) was also computed. The high posterior probability (P > 99%) of each site indicated that these sites have undergone positive selective pressure.
The amino acid composition in the selected site was detected by the M2a model in the 67 HA protein sequences and listed in Table II. In site 2T, threonine, alanine and valine are found in 57.1%, 39.3%, and 3.6% of the respective HA protein sequences. In site 3T, threonine and alanine were found in 41.6% and 58.4% of the respective HA protein sequences. In the site 197A, the amino acids with composition from high to low are valine, alanine, threonine, glutamic acid, and glycine. In site 233Q, the amino acids with composition from high to low are glutamine and leucine. In site 380R, the amino acids with composition from high to low are lysine and argnine, which are alkalescence amino acids.
| Sites | Residue | Percentage of residues % | Site ω (±SE) |
|---|---|---|---|
| 2 | T/V/A | 57.1/3.6/39.3 | 5.703 ± 1.030 |
| 3 | T/A | 41.6/58.4 | 5.615 ± 1.152 |
| 197 | T/V/A/G/E | 19.6/39.3/35.7/1.8/3.6 | 5.703 ± 1.030 |
| 233 | Q/L | 57.1/42.9 | 5.703 ± 1.030 |
| 380 | R/K | 44.6/55.4 | 4.287 ± 2.075 |
- The amino acids in the positive selective site from human infected isolates are showed in black boldface.
The amino acid composition of the selected sites from human isolates is listed in black bold type, involving threonine, glutamic acid, leucine, and lysine.
The amino acid mutation rate of AIV is similar to other influenza viruses due to the absence of an emending mechanism during RNA virus replication. There exist antigenic shift and drift ubiquitously in the evolution of AIV type A. The two changes occur predominantly in the HA or NA gene when there is a point of mutations in the HA gene. The HA protein will change its amino acid sequence and protein structure. The AIV would then escape from the recognition of the host immune system. Additionally, if there are mutations in the key amino acid sites of the HA protein, particularly in the receptor-binding site (RBS), the ability of transmission of the virus to a different host may be enhanced. In this article, only the subtype H9N2 was considered for study.
The 3D structure of the HA protein of the H9N2 virus is shown in Figure 3. The receptor-binding sites of H9N2, as shown in red in Figure 3, are composed of 134–138, 183, 190, and 224–228 site residues which are highly-conserved [Matrosovich et al., 2001; An et al., 2005]. Selected sites are shown in blue are in different positions from the receptor-binding sites.

3D structure of the H9N2 HA protein. Selected sites are shown in blue; receptor-binding sites are shown in red; cleavage sites are shown in black; HA1 strain are shown in green; HA2 strain are shown in light green; this figure of 3D structure of H9N2 HA protein is displayed by RasTop2.6.
Considerable research work has indicated that HA1 can be bound to the receptor of the host cell, and HA2 is an important subunit involved in cell membrane fusion [Plotkin et al., 2002; Saito et al., 2002; Li et al., 2005]. In Figure 3, all selected sites are in the HA1 strain (shown in green color), except the 380R site which is in the HA2 strain (shown in light green). This indicated that those residues involved with cell membrane fusion do not undergo selection. Those selection sites 2T, 3T, 14T, 165D, 197A, 233Q are not in the key location, but in the rim of RBS. This might affect the mutation of the receptor-binding site and even lead to the transmission of H9N2 to a different host. Enhanced surveillance should therefore be undertaken for the H9N2 subtype AIV to predict the key residue site mutations.
Protein Motif Detection
In order to analyze the HA protein domain and changes within the domain, the protein motif of 67 amino acid sequences were detected using the software OMIGA2.0. The results indicated that all sequences contain five motifs: asn_glycosylation, ck2_phospho, myristyl, pkc_phospho, and tyr_phospho, of which the modes are N-{P}-[ST]-{P}, [ST]-X(2)-[DE], G-{EDRKHPFYW}-X(2)-[STAGCN]-{P}, [ST]-X-[RK], and [RK]-X(2,3)-[DE]-X(2,3)-Y respectively. The results are shown in Table III.
| No. | asn_glycosylation | CK2_phospho | Myristyl | PKC_phospho | TYR_phospho | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bird | Human | Bird | Human | Bird | Human | Bird | Human | Birds | Human | |
| 1 | 29 | 29 | 16 | 16 | 57 | 57 | 147 | 147 | 149 | ○ |
| 2 | ● | 105 | 36 | 36 | 77 | 77 | 158 | 158 | 515 | 515 |
| 3 | 141 | 141 | ● | 195 | 81 | 81 | 163 | 163 | / | / |
| 4 | ● | 206 | ● | 213 | 111 | 111 | 220 | 220 | / | / |
| 5 | 218 | 218 | 204 | ○ | 146 | 146 | 250 | 250 | / | / |
| 6 | 298 | 298 | 409 | 409 | ● | 270 | 292 | 292 | / | / |
| 7 | 305 | 305 | 445 | 445 | 295 | 295 | 322 | 322 | / | / |
| 8 | 492 | 492 | 504 | 504 | 296 | 296 | 336 | 336 | / | / |
| 9 | 551 | 551 | / | / | 339 | 339 | 379 | 379 | / | / |
| 10 | / | / | / | / | 342 | 342 | 387 | 387 | / | / |
| 11 | / | / | / | / | 350 | 350 | 489 | 489 | / | / |
| 12 | / | / | / | / | 354 | 354 | 521 | 521 | / | / |
| 13 | / | / | / | / | 369 | 369 | 553 | 553 | / | / |
| 14 | / | / | / | / | 552 | 552 | / | / | / | / |
- ●, represents the absent motif site in isolates from infected poultry compared with human isolates; ○, represents the absent motif site in human-infected isolates compared with poultry-infected isolates.
- The above protein motifs are detected using software OMIGA2.0.
The potential glycosylation sites of the HA protein can affect the host cell type and its binding capacity. This is one of the main factors affecting the virulence of H9N2 AIV. Poultry isolates in China contain the common glycosylation sites. The two isolates from human cases in Hong Kong in 1999 contain the glycosylation sites in 105 and 206, except for the same glycosylation sites in 29, 141, 218, 298, 305, 492, and 551 with the poultry-infected isolates. It is suggested that the glycosylation level of the 105 and 206 sites should be kept under surveillance to monitor virulence changes in the H9N2 AIV.
Protein phosphorylation is an important post-translational modification process and plays an important role in cell differentiation and cell signal transduction. Most isolates from infected poultry contain the common 6 Casein Kinase II (CK2) phosphorylation site and 2 tyrosine phosphatase sites. Compared with human isolates, there is an absence of two CK2-phosphorylation sites at 195 and 213 amino acids and an extra tyrosine phosphatase site in at 149 in poultry-infected isolates. All 67 AIV isolates have the same 13 PKC_phosphate sites. All of the above results are shown in Table III. To determine whether these characteristics are involved in overcoming the species barrier deserves further research.
The sequence pattern in the HA proteolytic cleavage site is also a primary determinant of the virulence of the virus. Generally, the amino acids at the HA protein cleavage site are mostly alkaline amino acids, which are recognized and cleaved by proteases in the host cell. All HPAI viruses have many alkaline amino acids in adjacent cleavage sites of the HA protein. The 67 AIV isolates in China have the sequence pattern -PARSSRGLF- in the cleavage site, which is compatible with LPAI viruses, but different from the highly pathogenic AIV sequence pattern: -PARKKKKRGLF-.
Glycosylation sites in adjacent cleavage sites may affect the HA precursor protein cleaved by proteases. The HA proteins in 67 AIV isolates contain 7 potential glycosylation sites, which are far from or close to the cleavage site. Whether these sites can change the cleavage of the HA protein requires further research.
DISCUSSION
Although H9N2 is a LPAI virus, it has the capability of transformation and reassortment with other subtype AIVs, and can transform into a variant or a novel HPAI virus. Among the eight segmented single strand RNAs, HA protein is a glycoprotein on the virus surface, which can induce antibody response in the cell. The HA protein is a primary protection antigen in the AIVs, therefore the amino acid site analysis and evolution of the HA gene is meaningful for vaccine and preventative measures.
Phylogenetic Tree and Recombination Analysis
All 68 complete sequences of H9N2 AIV HA protein isolated during 1994–2007 were analyzed by various methods. The phylogenetic analysis results show that the early isolates between 1994 and 1999 in China are dispersed and belong to a Euro-Asia sublineage, and are in a different evolutionary direction from the human isolates in Hong Kong in 1999. The fact that two H9N2 influenza viruses were isolated from domestic pigs in Hong Kong in early 1998 indicated that avian-to-mammalian transmission of H9N2 AIV had already occurred in China. In mid 1998, five humans were infected with H9N2 influenza virus in the Guangdong province, and they recovered from the disease [Guo et al., 1999]. In March 1999, there emerged the first H9N2 AIV human death in Hong Kong [Peiris et al., 1999b]. These events indicated that the H9N2 AIV had broken the species barrier and begun infecting humans, and had the possibility of becoming another potential pandemic HPAI subtypes.
Previous studies have indicated that intergenic reassortment occurs in the evolutionary process of segmented AIV [Guan et al., 1999]. Human H5N1 viruses in Hong Kong in 1997 were reassortants acquiring their internal genes from the A/quail/HK/G1/97-like H9N2 virus. It is known that intragenic recombination occurs in the segmented RNA viruses. To date, except the Chile 2002 H7N3 HPAI virus with an HA gene recombination with other nucleoprotein, and the Canada 2004 H7N3 HPAI was a virus with HA gene recombination with other matrix gene [Alexander, 2007], no other reports provide evidence that intragenic recombination exists in AIV [Chare et al., 2003; Boni et al., 2008]. By recombination detection using RDP3.0, only the Ck/YN/nh/01 virus was detected as recombinant sequences of Dk/NJ/01/99 and Ck/GX/9/99. In order to ensure the success of analysis, this isolate was excluded from the study of positive selection.
Comparison of Selected Sites on HA Protein in Different Epidemic Periods
There are in total seven positive selection sites in the HA protein detected by the software Codeml. All the selection sites except for the 380R site are on the HA1 peptide chain, which has the primary antigenic sites and receptor-binding sites. Although seven detected selection sites are not the antigenic sites or receptor-binding sites, some of these sites are still on the rim of RBS. Therefore, the surveillance of H9N2 AIV HA protein should be strengthened to predict mutation of its key sites. By investigating the selection for three groups (1994–1997, 1998–1999, and 2000–2007 epidemic groups) using PAML, the results indicated that the evolution of H9N2 is currently a slow process.
The positively selected sites in three epidemic groups of 1994–1997 group, 1998–1999 group, and 2000–2007 group were all identified using the Codeml program. All three selection models (M2a, M3, and M8) showed that selection occurred in three epidemic groups (Table IV), however the percentage of selected sites in the HA protein from three epidemic groups were different. For example, 0.6–1.3% of the sites in 1994–1997 epidemic group were under positive selection with ω values between 4.37 and 10.02, 0.2–1.1% of the sites in 1998–1999 epidemic group are under positive selection with ω values between 3.51 and 8.53, and 0.6–0.9% of the sites in 2000–2007 epidemic group are under positive selection with ω values between 5.04–7.04. These results indicated for the first time that the evolution of the HA protein was still under positive selection at particular sites, and more active evolution in the 1994–1997 groups and 1998–1999 groups. The positive selection sites are changeable in different epidemic groups. These observations provide evidence for understanding the molecular adaptation of H9N2. In recent years, especially during 2000–2007, the HA protein was not under strong selection, so attention should be paid to its evolution due to the intergeneic recombination.
| Epidemic phases | Model | LnL | dN/dS | Estimated parameters | 2Δl | Positive selection |
|---|---|---|---|---|---|---|
| 1994–1997 epidemic group | M0(one-ratio) | −3,584.44 | 0.27 | ω = 0.27 | 72.54 | None |
| M3(discrete) | −3,548.17 | 0.30 | p0 = 0.92823, p1 = 0.06537, p2 = 0.00640 | (13.28) | 3T,14T,65P,91G,165D,197A,204R | |
| ω0 = 0.11812, ω1 = 1.88467, ω2 = 10.02438 | 233Q,255N,359Y,371M,380R,410V,412T,468M | |||||
| M1(NearlyNeural) | −3,562.23 | 0.24 | p0 = 0.83583, p1 = 0.16417 | 25.36 | None | |
| M2a (PositiveSelection) | −3,549.55 | 0.29 | p0 = 0.96644, p1 = 0.00089, p2 = 0.03267 | (9.21) | 3T,14T,65P,91G,197A,204R | |
| ω0 = 0.15184, ω1 = 1.00000, ω2 = 4.37466 | 233Q,359Y,380R,410V | |||||
| M7(beta) | −3,558.41 | 0.23 | p = 0.01294, q = 0.04091 | 19.74 | None | |
| M8(beta and ω) | −3,548.54 | 0.30 | p0 = 0.98707, p1 = 0.01293 | (9.21) | 3T,14T,65P,91G,165D,197A,204R | |
| p = 0.17971, q = 0.69803, ω = 7.15656 | 233Q,359Y,380R,410V,412T | |||||
| 1998–1999 epidemic group | M0(one-ratio) | −5,595.88 | 0.22 | ω = 0.22 | 124.44 | None |
| M3(discrete) | −5,533.66 | 0.24 | p0 = 0.79766, p1 = 0.19434, p2 = 0.00800 | (13.28) | 2T,197A,233Q | |
| ω0 = 0.06183, ω1 = 0.81264, ω2 = 4.00322 | ||||||
| M1(NearlyNeural) | −5,536.59 | 0.23 | p0 = 0.83540, p1 = 0.1646 | 5.52 | None | |
| M2a (PositiveSelection) | −5,533.83 | 0.24 | p0 = 0.83518, p1 = 0.16266, p2 = 0.00216 | (9.21) | 2T,197A,233Q | |
| ω0 = 0.07383, ω1 = 1.00000, ω2 = 8.53487 | ||||||
| M7(beta) | −5,538.60 | 0.22 | p = 0.16377, q = 0.57596 | 9.24 | None | |
| M8(beta and ω) | −5,533.98 | 0.24 | p0 = 0.98916, p1 = 0.01084 | (9.21) | 2T,14V,39N,62N,78I,86Q,89G,120S | |
| p = 0.21864, q = 0.85675, ω = 3.51097 | 148K,165N,197A,223M,233Q,305V,380R | |||||
| 2000–2007 epidemic group | M0(one-ratio) | −5,303.06 | 0.16 | ω = 0.16 | 128.82 | None |
| M3(discrete) | −5,238.65 | 0.18 | p0 = 0.58033, p1 = 0.40986, p2 = 0.00981 | (13.28) | 2T,3T,197V,233Q,281N | |
| ω0 = 0.00653, ω1 = 0.31888, ω2 = 5.04282 | ||||||
| M1(NearlyNeural) | −5,253.80 | 0.16 | p0 = 0.91991, p1 = 0.08009 | 25.76 | None | |
| M2a (PositiveSelection) | −5,240.92 | 0.19 | p0 = 0.92795, p1 = 0.06582, p2 = 0.00624 | (9.21) | 2T,3T,197V,233Q,544L | |
| ω0 = 0.09065, ω1 = 1.00000, ω2 = 7.03982 | ||||||
| M7(beta) | −5,258.03 | 0.17 | p = 0.18471, q = 0.90750 | 38.66 | None | |
| M8(beta and ω) | −5,238.70 | 0.18 | p0 = 0.99114, p1 = 0.00886 | (9.21) | 2T,3T,197V,233Q,281N,282S,380K | |
| p = 0.38184, q = 2.31058, ω = 5.41252 |
- The posterior probability of black boldface site is over 99%, the other is over 95%. The values in parentheses represent the significant level of 0.01 with a χ2 distribution at d.f. = 4 (M0 vs. M3) or 2 (M1a vs. M2a and M7 vs. M8). M0, M1, M2a, M3, M7, M8 are site models which allow the ω ratio to vary among sites respectively. Above results were obtained by codeml from PAML software package version 3.15.
The LRT statistical results revealed that three selection models fit the data better than three null models in the three epidemic groups, which supports further the presence of amino acid sites under selection pressure in the HA protein (except that M2a model is not significant in 98–99 epidemic groups under the level of 0.01).
The protein motifs are also detected by OMIGA2.0, and it was found that there are differences in the motifs between infecting isolates of poultry and isolates infecting humans. This indicates that the H9N2 overcoming species barrier to infect humans might be involved with mutation in the glycosylation sites which should be also an enhanced surveillance.
ADDITIONAL NOTES ON THE MATERIALS AND METHODS USED IN THIS STUDY
The complete or nearly complete-length sequences of HA gene in AIV isolates from China between 1994 and 2007 were retrieved from Genbank (only one representative sequence was selected from those 100% similar alignment sequences, and from the same geographic site and same time. Those containing unknown residues were deleted). In total, 68 HA gene sequences of the AIVs were obtained for studied in this article, of which the accession numbers are as follows: AF384557, AF536689, AF461526, AF536690, AF536692, AF536693, DQ064360, DQ064362, DQ064374, DQ064377, AF461527, AF508564, AF508566, AF508569, AF508570, AF508572, AF508573, AF156373, AF156374, AF156375, AF461509, AY043015, AY043017, AY043018, AY043019, DQ064364, DQ064370, DQ064375, DQ064379, DQ681221, EF070733, AF461510, AF461511, AF461512, AF461516, AF461517, AF461521, AF461522, AF461528, AF461532, AF508562, AF508565, AF508568, AF508571, AJ404626, AJ404627, AY206676, AY206677, DQ981554, AF461515, AF461518, AF461524, AY294658, AY364228, DQ681203, DQ681207, EU216085, EU216087, EU216092, DQ997465, DQ681216, DQ064357, DQ064369, DQ465400, EU086226, EU086303, AY664665, and AY664670.
Methods
Two phylogenetic trees from a total of 68 HA gene sequences were constructed by Seqboot.exe, Distance.exe, and Neighbor.exe from the PHYLIP software [Felsenstein, 1989]. The reliability of the trees was evaluated by the bootstrap method with 100 replicates. The dN/dS value was used to detect positive selection. Since gene recombination can result in false high dN/dS values and a false positive selection. RDP3.0 software [Martin and Rybicki, 2000] was employed to detect the intragenic recombination between 1994 and 2007 epidemic H9N2 viruses in China.
It is known that different amino acid sites have different biological functions and are subject to different evolutionary selection pressures. Therefore nonsynonymous substitutions were compared with synonymous substitutions per site to recognize selective pressure based on amino acid sequences. For nucleotide or amino acid sequences, the estimation rate ratio  of nonsynonymous substitution rates dN and synonymous substitution rates dS can be used to detect the selection pressure of evolution [Yang et al., 2000]. The ω is an important factor in understanding the dynamics of molecular sequence evolution. The ω > 1 indicates that nonsynonymous substitution is predominant in the amino acid evolution process, gradually stabilized in the amino acid sequence with high probability, and the gene underwent positive selection; ω = 1 indicates that the gene has undergone random drift and negative (purifying) selection. However, during the evolutionary process, because of the complex protein structure, the selection pressure in some amino acid sites is significantly different from that of the whole sequence. There are no reports regarding the site selection pressure of H9N2 AIV subtype HA protein. This motivated us to analyze HA protein site selection pressure.
of nonsynonymous substitution rates dN and synonymous substitution rates dS can be used to detect the selection pressure of evolution [Yang et al., 2000]. The ω is an important factor in understanding the dynamics of molecular sequence evolution. The ω > 1 indicates that nonsynonymous substitution is predominant in the amino acid evolution process, gradually stabilized in the amino acid sequence with high probability, and the gene underwent positive selection; ω = 1 indicates that the gene has undergone random drift and negative (purifying) selection. However, during the evolutionary process, because of the complex protein structure, the selection pressure in some amino acid sites is significantly different from that of the whole sequence. There are no reports regarding the site selection pressure of H9N2 AIV subtype HA protein. This motivated us to analyze HA protein site selection pressure.
The site selection pressure of the HA protein in H9N2 was analyzed by the program codeml in the PAML3.15 Software Package [Yang, 1997] which is based on the Maximum Likelihood method. Six site models, M0(one-ratio), M1a(NearlyNeutral), M2a (PositiveSelection), M3(discrete), M7(beta), and M8(beta and ω) were used in the detection [Yang et al., 2000, 2005]. M0 assumes all sites with same ω ratio. M1a assumes two classes of sites in proteins with 0 < ω0 < 1 (percentage p0) and ω1 = 1 (percentage p1). M2a adds an extra class of sites which w2 is estimated from the data. M3 uses three site classes with the ω ratios (ω0, ω1, and ω2) estimated from the data. M7 assumes a beta distribution (p, q) for 10 different ω ratios in the interval (0, 1). M8 adds a class of sites with positive selection (ω > 1) to the beta (M7) model.
The Likelihood ratio test (LRT) is performed for detecting the significance of positive selection sites. Three LRTs (M0 vs. M3, M1a vs. M2a, and M7 vs. M8) are used to infer the significance of positive selection sites detection. The sites with high posterior probabilities (P > 0.95) coming from the class with ω > 1 are believed to be under positive selection.
The protein motifs of 67 amino acid sequences were detected using the software OMIGA2.0 by scanning all the 67 amino acid sequences of H9N2 subtype.
Acknowledgements
The study was supported by grants from Natural Science Foundation of Jiangsu Province in China (BK2010500).

