

Merging molecular imaging and RNA interference: Early experience in live animals
Abstract
The rapid development of non-invasive imaging techniques and imaging reporters coincided with the enthusiastic response that the introduction of RNA interference (RNAi) techniques created in the research community. Imaging in experimental animals provides quantitative or semi-quantitative information regarding the biodistribution of small interfering RNAs and the levels of gene interference (i.e., knockdown of the target mRNA) in living animals. In this review we give a brief summary of the first imaging findings that have potential for accelerating the development and testing of new approaches that explore RNAi as a method for achieving loss-of-function effects in vivo and as a promising therapeutic tool. J. Cell. Biochem. 104: 1113–1123, 2008. © 2008 Wiley-Liss, Inc.
Post-transcriptional gene silencing has been associated with diverse regulatory processes such as transposon silencing, antiviral defense mechanisms, gene regulation, and chromatin modification [Zamore and Haley, 2005]. Because of high specificity in nature, the small interfering RNA (siRNA) technology, which is based on the effect of gene silencing, is believed to result in efficient therapeutic approaches that will enable safe down-regulation of the expression of genes associated with various disease states [McManus and Sharp, 2002]. The use of siRNA is especially promising for targets that are otherwise not amenable to traditional therapies (known as non-druggable targets) [Soutschek et al., 2004].
The discovery of efficient double-stranded RNA (dsRNA) interference with gene expression in nematode C. elegans [Fire et al., 1998] prompted siRNA genome-wide screens in this organism [Kamath and Ahringer, 2003] due to the ease of internalization and utilization of siRNA by nematodes [Hull and Timmons, 2004]. However, for a while similar success evaded investigators that attempted to induce long dsRNA-mediated knock-down of target genes in mammalian cells. The main reason for failure was the activation of antiviral response to dsRNA and sequence-nonspecific interferon-mediated mRNA degradation effects [Stein et al., 2005]. However, the discovery of the mechanism underlying the processing of longer dsRNA into the small 21–22 nt dsRNA segments using Drosophila in vitro system [Zamore et al., 2000] enabled critically important experiments that lead to a successful target-specific gene silencing in mammalian cells. The initial experiments proved that in commonly used cell lines (293, COS etc.) 21–22 nt-long dsRNA with overhanging 3′-ends allowed efficient knock-down of target marker genes [Elbashir et al., 2001]. The delivery of these short siRNA into the cells and the demonstration of silencing effects was achieved by using simple co-transfection of siRNA molecules together with reporter plasmids carrying firefly and Renilla (sea pansy) luciferase cDNAs [Elbashir et al., 2001].
Double-stranded synthetic siRNAs that were used in the above study can be synthesized using oligonucleotide automated synthesis or, alternatively, can be produced using in vitro transcription and further Dicer RNAseIII processing [Banan and Puri, 2004] (Fig. 1). These small siRNA or dsRNA generated from micro RNA (miRNA) are incorporated into the multiprotein RNA-induced catalytic silencing complex (RISC), where a complex of antisense strand of siRNA duplex and small RNA-binding argonaute protein (argonaute-2 in mammals) are mediating interaction and degradation of the cognate mRNA target due to the recruitment of endonucleases in a specialized cytoplasmic “silencing” compartment [Zamore and Haley, 2005; Joshua-Tor, 2006] (Fig. 1). This allows increasing the efficacy of RNA interference (RNAi) in mammalian cells that otherwise is rate-limited and depends exclusively on Dicer RNAse III activity. However, synthetic siRNA delivery does not usually permit long-term silencing effects. Alternatively, the use of polymerase III promoters directing the transcription of human RNA H1 and small nucleolar RNA U6 genes [Carbon et al., 1987; Myslinski et al., 2001] enable transcription of small hairpin RNA (shRNA) directly from expression vector cassettes. Lentiviral transduction of shRNA results in long-term knock-down of target genes in endothelial cells and mouse brain [Makinen et al., 2006]. U6 promoter was reported as more efficient than H1 in GFP silencing in vitro, leading to 80% GFP knockdown at an average of one integrated vector genome per target cell genome [Makinen et al., 2006]. Earlier studies suggested that translating RNAi technology to mouse embryo stem cells for generating transgenic animals with targeted gene downregulation is not straightforward due to low efficacy of promoters and lethality due to interferon response [Cao et al., 2005]. However, there is a critical difference between minimal 200 bp H1 promoter and much longer genomic fragments containing this promoter. The use of the latter improved the outcome of embryonic stem cell experiments [Berlivet et al., 2007]. Furthermore, lentiviral H1-promoter driven siRNA expression can be made inducible by stuffer reporter deletion thereby potentially decreasing non-specific toxic effects [Heinonen et al., 2005]. Since a long-term knockdown of target mRNAs by using Pol III promoters is problematic [Fish and Kruithof, 2004], conditional Pol II-driven expression of microRNA mimics was suggested as an alternative for achieving long-term inducible RNAi [Silva et al., 2005; Shin et al., 2006]. Despite the evidence of impressive progress in applying siRNA technology in mammalian cells and highly efficient target gene knock down, there are still unresolved problems associated with RNAi technology: (1) the delivery of siRNA and shRNA to cells in vivo; (2) the potential for interferon response, which is not eliminated completely by using short interfering RNA; (3) the off-target silencing effects. Some of these problems are tackled at the level of experiments in living animals. In these experiments the aspects related to RNAi delivery and noninvasive imaging of RNAi effect are gaining more prominence.

Simplified RNAi pathways in mammalian cell (adapted from Zamore and Haley 2005). Exogenous RNAi that are most commonly used for gene silencing in vivo are highlighted in red. Both miRNA and shRNA expression can be transcribed from exogenous DNA vector molecules that need to reach the nucleus. Micro RNA precursors are processed by Drosha RNAse complex, shRNA do not require the processing. The resultant small hairpin RNAs are exported in the cytoplasm and processed further by Dicer RNAse complex. Association with Argonaute-2 (Ago-2) exonuclease of RISC (RNA-induced silencing complex) results in antisense-guided interaction with target mRNA and a formation of silencing complex in specialized cytoplasmic bodies where target mRNA undergoes decay. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
APPROACHES TO siRNA DELIVERY
As discussed above, the knock-down efficacy of RNAi depends on whether the RNA duplex will eventually reach the cytoplasm of the target cells. In the case of chemically synthesized siRNA duplexes the delivery to cytoplasm is usually considered sufficient because they do not have to be further processed by nuclear RNAse III (e.g., RNAse Drosha that processes pri-miRNAs, Fig. 1). Some artificial and dumbbell-like shRNA precursors [Seyhan et al., 2006] seem to be amenable to Dicer cleavage and possibly do not require Drosha for further processing into small RNA duplexes. However, in the case of larger shRNA-encoding DNA vectors the access to cell nucleus is essential since transcriptional machinery is required for the synthesis of shRNA. It is thus inevitable that the existing barriers to RNAi delivery would severely limit the efficacy of both synthetic siRNA, as well as shRNA encoding vectors. Many initial attempts to knock-down gene expression in mammals by applying systemic administration routes showed a total lack of effect [Lewis and Wolff, 2007]. However, the delivery of both siRNA and shRNA constructs in vivo can be achieved through an approach involving “pressure” (i.e., bolus) injection of large volumes of siRNA solutions (approximately 9% wt/vol of total animal blood volume [McAnuff et al., 2007]). This method was developed initially for delivering “naked” plasmid vectors into mouse liver resulting in specific expression of transgenes in hepatocytes [Lewis and Wolff, 2007]. Bolus injections performed using lower volumes or within longer times result in poor in vivo transfer [Lewis and Wolff, 2007]. There are obvious disadvantages of the above delivery approach, which limits its use to simplified model studies in rodents. However, since both gene expression and gene silencing constructs could be co-delivered using rapid and simple procedure, this technique is essential for comparing and optimizing silencing constructs [McAnuff et al., 2007; Bartlett and Davis, 2007b].
Since regular intravenous administration of siRNA is inefficient, two other approaches were experimented with by several groups. The first utilizes electroporation and electric pulse delivery [Eefting et al., 2007; Golzio et al., 2007], and the second involves incorporation of siRNA in a variety of particles, for example, positively charged iron oxide nanoparticles [Medarova et al., 2007], liposomes [Bisanz et al., 2005; Kim et al., 2007] or copolymer-based nanoparticles [Bartlett et al., 2007; Rozema et al., 2007]. The nanoparticle approach permits condensation of chemically synthesized or in vitro transcribed siRNA molecules in attempt to improve their ability to traverse cell membranes [Kim et al., 2007; Medarova et al., 2007]. In many cases, the incorporation of siRNA into particles and ribonucleoprotein complexes involves the use of chemical modification of 3′-end of the sense siRNA strand with membrane-tropic moieties including cholesterol [Soutschek et al., 2004] or fatty and bile acids [Wolfrum et al., 2007]. Hydrophobic moieties result in recombination of modified siRNA with HDL and LDL particles and increased liver tropism. Alternatively, special liver-targeted ApoB siRNA-binding polymer delivery systems were designed to release the contents of nanoparticles in the endosomes [Rozema et al., 2007]. The latter undergo acidification that supposedly results in “uncoating” of siRNA-polymer complexes, destabilization of endosomes and the release of siRNA in the cytoplasm. Thus, the problems associated with cytoplasmic delivery of dsRNA are very similar to those that were dealt with 10–15 years ago when nonviral gene delivery vectors were formulated into sophisticated transfection complexes with the attempt to overexpress therapeutic proteins in vivo [Ledley, 1994]. However, the field is clearly benefiting from the lessons learned during that period.
ASSESSMENT OF RNAi DELIVERY USING IMAGING
Determining the biodistribution of siRNA and shRNA constructs is extremely important in identification of target and nontarget organs of potential RNAi therapies and for predicting potential toxicities to these organs. In most cases, the use of animal experiments enables the comparison of the above RNAi delivery methods. These experiments require covalent “tagging” of oligoribonucleotides. For example, oligoribonucleotides can be labeled by tritium at C8 positions of purines using heat-exchange method [Mook et al., 2007]. In the recent detailed study of the influence of protein binding and protein-based delivery of chemically modified ApoB siRNA (i.e., the small interfering siRNA that silences ApoB expression), Wolfrum et al. 2007 utilized 5′-phosphorylation of siRNA with γ-AT(32P). Tracking of 32P-siRNA or 3H-siRNA and its metabolites requires scintillation counting of tissues removed from sacrificed animals or, alternatively, relies on experiments with perfused isolated organs. The alternative to 32P-labeling is noninvasive imaging which is ideally suited for determining biodistribution of constructs used for gene delivery and transfer in vivo (reviewed in Bogdanov and Weissleder 1998 and Bogdanov and Weissleder 2002). For example, both major radionuclide-based tomographic imaging modalities, positron emission tomography (PET) and single-photon computed tomography (SPECT), can potentially be used for imaging siRNA biodistribution in live animals. There are many similarities between both radionuclide imaging methods, though PET requires a very high energy emitter but less injected radioactivity dose and usually provides better quantitation and temporal resolution than SPECT. SPECT, on the other hand, uses “safer” radioactivity emitters and usually allows to achieve better spatial resolution (less than 1 mm). By “fusing” radionuclide images with anatomic CT images (a computer-aided process known as “registration”) one can potentially obtain a 3-D map of radioactivity distribution within a given organ.
The initial experience with radionuclide imaging of RNA labeled with gamma-emitting isotopes suggested that radionuclide imaging can serve as an alternative to conventional radioisotope experiments [Liu et al., 2007]. To test the use of planar image acquisition approach (i.e., without tomographic image reconstructions) Liu et al. used stabilized 18-nt 2′-O-methyl oligoribophosphorothoates with 5′-amino linker conjugated with hydrazine nicotinamide acid (HYNIC). The latter is a chelating molecule capable of efficiently binding reduced pertechnetate-99m (99mTcO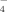 ). Technetium-99m emits 140.5 keV gamma photons that can be detected in live animals by using standard gamma cameras with NaI detectors. The authors observed predominant accumulation of 99mTcHYNIC labeled RNA analog in the liver (about 8–9% dose) and kidneys (13–26% dose). However, instead of duplex siRNA the authors utilized either sense, or antisense strands of RNA, thus, no conclusions regarding the distribution of RNA duplexes could be made [Liu et al., 2007]. The fast kidney filtration is typical for all siRNA since the duplexes are small, and retention in the liver is probably due to scavenging of phosphorothioates by endothelial cells of the liver as in the case of oligodeoxyphosphorothioates [Bijsterbosch et al., 1997].
). Technetium-99m emits 140.5 keV gamma photons that can be detected in live animals by using standard gamma cameras with NaI detectors. The authors observed predominant accumulation of 99mTcHYNIC labeled RNA analog in the liver (about 8–9% dose) and kidneys (13–26% dose). However, instead of duplex siRNA the authors utilized either sense, or antisense strands of RNA, thus, no conclusions regarding the distribution of RNA duplexes could be made [Liu et al., 2007]. The fast kidney filtration is typical for all siRNA since the duplexes are small, and retention in the liver is probably due to scavenging of phosphorothioates by endothelial cells of the liver as in the case of oligodeoxyphosphorothioates [Bijsterbosch et al., 1997].
Instead of HYNIC, DOTA (a macrocyclic chelating molecule) can be covalently linked to 5′-amino linker on siRNA strand. DOTA chelates a variety of gamma-photon as well as positron-emitting metal cations. One recent study included a parallel investigation of biodistribution and gene knock-down induced by siRNA labeled with a positron emitter (64Cu) [Bartlett et al., 2007] (Fig. 2A). The use of PET enabled dynamic imaging of siRNA, as well as nanoparticle complexes of siRNA in various organs, including tumor xenografts with time scale resolution of 5 min. The authors were able to demonstrate that nanoparticles obtained by condensing siRNA molecules with polycations were accumulating in tumors at the same levels regardless of whether they were targeted to transferrin receptors or stabilized by polyethylene glycol [Bartlett et al., 2007]. This important observation helped in interpreting the results of imaging of target gene expression marker, that is, luciferase.

A: Multimodality in vivo imaging of siRNA nanoparticle delivery and RNAi effect using micro-PET/CT and bioluminescence imaging. Top row-fused micro-PET/CT images showing tumor-associated (arrow) activity 1 day after injection of targeted (Tf) and nontargeted (PEG) nanoparticles containing 64Cu-DOTA-siRNA. Bottom row-luminescence images of the same mice shown above before injection and 1 day after injection. Graph—relative change in luciferase expression 1 day after injection of Tf-targeted (Tf) and nontargeted (PEG) nanoparticles containing 64Cu-DOTA-siRNA for simultaneous PET imaging. Reprinted with permission from Bartlett et al. 2007. B: Luminescence imaging of firefly luciferase expression knock-down by specific shRNA expression, which was co-delivered by pressure (bolus) intravenous co-injection of MDR1-luc fusion protein expression plasmid (pMDR1-Fluc, 1 µg), Renilla luciferase expression plasmid (pRLuc-N3, 1 µg; as transfection control) and the corresponding shRNA expression vector (10 µg). Bioluminescence images of Rluc expression imaged by using coelenterazine (Renilla luciferase substrate, top) and P-glycoprotein-FLuc expression with D-luciferin (firefly luciferase substrate, bottom). Mice co-injected with 10-fold excess of control (left), scrambled shRNAi (middle), or shRNAi against MDR1 (right). Reprinted with permission from Pichler et al. 2005. C: In vivo near-infrared optical imaging of mice that had bilaterally implanted engineered rat glioma tumors (9L-GFP and 9L-RFP) before and 48 h after intravenous injection of siRNA complexes with positively charged iron oxide nanoparticles. siRNA was designed to knock-down GFP expression (phGFP-S65T nucleotides 122–141: 5′-GCA AGC TGA CCC TGA AGT TC-3′) but not was not inhibiting RFP expression. Imaging showed a marked decrease in 9L-GFP fluorescence (P = 0.0083) but not in 9L-RFP fluorescence levels. To generate GFP/RFP reconstructions, GFP and RFP images were acquired separately and then merged. Reprinted with permission from Medarova et al. 2007. D: Imaging of luciferase activity in mice that were implanted with U251-HRE glioma cells in the flank. The cells expressed luc under control of HRE (hypoxia-responsive element). Mice were imaged on day 15 of thrice weekly intratumoral injection time course. Mice on the left were injected with siRNA1589 (designed to knock-down HIF-1alpha), whereas those on the right were injected with the scrambled siRNA control. The graph quantifying the average radiance (normalized) of the glioma (U251-HRE) tumor cells imaged during the siRNA injection time course is shown below. Day 0 is the baseline luciferase activity before siRNA injection. The luciferase activity is significantly less in the group injected with siRNA1589 by day 15 (*P = 0.037, n = 5 mice/group). Reprinted with permission from Gillespie et al. 2007. E: Bioluminescence imaging of synergistic effect of siRNA and shRNA in mice. Mice were dosed in triplicate with 0.1 µg of pGL3 and either 1 µg of pSEAP (a control irrelevant plasmid), pShagLuc (shRNA expression vector), siLuc1 (siRNA construct), control, or 0.5 µg of pShagLuc and siLuc1. The imaging signals (Y-axis) were translated in to mass units of expressed enzyme (YY-axis) and demonstrate a statistically significant synergistic effect for the combined dose siLuc1 and pShagLuc in vivo. P < 0.05 relative to pGL3 + pSEAP and P < 0.05 relative to pGL3 + pShagLuc. Reprinted with permission from McAnuff et al. 2007.
It is highly likely that future experiments will involve better characterized siRNA—based complexes with more acceptable positron-emitters (e.g., 18F labeled siRNA analogs for PET imaging). Fluorine-18 labeled oligonucleotides have been already tested as labeled imaging probes in vivo [Boisgard et al., 2005; Dolle, 2007]. With the development of more quantitative in vivo imaging experiments the current animal studies involving siRNA could be either complemented with, or replaced entirely by survival (and longitudinal) imaging studies. Below are described other examples of such initial studies, which were focused on establishing the degree of the target gene down-regulation.
THE USE OF IMAGING IN RNAi EFFICACY EXPERIMENTS
Imaging is very frequently used to detect gene knock-down by RNAi in cell culture. Imaging on the cellular level is essential for performing high-content and high-throughput screening of target gene loss-of-function with RNAi libraries using automated standard or time-lapse fluorescence microscopy [Rines et al., 2006]. Recent examples include shRNA lentiviral libraries [Moffat et al., 2006] and siRNA transfection libraries printed in microarrays in cell-imaging chambers [Neumann et al., 2006]. These screens are capable of targeting about 22,000 mouse and human gene transcripts. Targeting of 1,028 genes of human mRNAs with multiple lentiviral constructs revealed new gene targets that are involved in regulation of mitotic progression [Moffat et al., 2006].
The validation of discovered RNAi targets does not usually require in vivo imaging of target gene down-regulation in living animals. However, the monitoring of anti-proliferative effects of RNAi over time is greatly assisted by imaging cellular proliferation indirectly, that is, by following the levels of marker gene expression in living animals. The choice between various gene expression marker proteins needs to made with caution since high stability of the protein product of gene expression can prevent early observation of RNAi effects. As a rule, the imaging marker proteins (i.e., knock-ins) are luminescent: due to the ease of optical imaging and the lack of appreciable background signal in native animals, the majority of in vivo studies are performed by measuring intensity of emitted luminescent light output of luciferases using CCD cameras (Table I). The use of fluorescent proteins (e.g., green fluorescent and red fluorescent proteins) is also feasible though the intrinsic autofluorescent signal in tissues in GFP channel can require spectral filtering of the background signal [Mansfield et al., 2005]. If given siRNA or shRNA are designed to knock-down the expression of marker proteins, in vivo imaging would reflect the efficacy of intracellular transfer of siRNA. If RNAi target and expression marker are expressed as separate mRNAs, imaging could potentially reflect the relative importance of the target transcript for cell proliferation, because the expression of marker genes will cease or will be down-regulated if RNAi construct does affect the proliferation-relevant target. Due to the difficulties in delivering shRNA expression vectors in vivo, in the majority of cases shRNA vectors are transfected or transduced using viral vectors into the cells in vitro and then these cells with altered patterns of gene expression are implanted in live animals (Table I). However, the bolus technique, which employs a co-injection of shRNA constructs and the vectors encoding the target sequence was also efficient and allowed the imaging of changes in the marker gene expression [McAnuff et al., 2007; Mook et al., 2007]. It has been initially demonstrated that chimeric protein translated from a single open reading frame and consisting of a fusion of target and marker proteins allows the use of the “marker” portion of the sequence as a template for readout of RNAi effect [Pichler et al., 2005]. By fusing MDR1 cDNA with Renilla or firefly luciferase, the shRNA effect directed at MDR1 gene product (P-glycoprotein) was monitored in vivo by measuring the change in light output of luciferase [Pichler et al., 2005] (Fig. 2B). The authors elegantly used the fact that the substrate of Renilla luciferase is pumped out of the cells by P-glycoprotein. Therefore, tumor cells that had P-glycoprotein knocked down by shRNA developed tumors that were more luminescent in vivo after the injecting the substrate than the control cells.
| Target gene | Pathology | Imaging modality | RNA interference mode | Animal model | RNAi delivery mode | References |
|---|---|---|---|---|---|---|
| Luciferase, TLR4 | N/A | BLI | shRNA | Mouse calf mucle | Local, electroporation |
Eefting et al. 2007 |
| Luciferase | N/A | BLI | siRNA | Mouse skin | Local, co-injection with Luc reporter |
Wang et al. 2007 |
| Luciferase | N/A | BLI or PET | (64Cu)DOTA-siRNA nanoparticles, transferrin-targeted | Human tumor xenograft | IV injection |
Bartlett et al. 2007 |
| Luciferase | BLI | Stabilized siRNA, coinjection with target plasmid | Mouse liver | Pressure injection |
Bartlett and Davis 2007b |
|
| Luciferase | Hepatitis B | BLI | siRNA ApoA-I/DOTAP/Chol complex | Mouse infected with HBV, Liver | IV injection |
Kim et al. 2007 |
| Luciferase | N/A | BLI | siRNA, pShagLuc | Normal mice | IV, pressure injection |
McAnuff et al. 2007 |
| Green fluorescent protein | N/A | FI | LNA-modified siRNA | Mouse, pancreatic adenocarcinoma xenografts | Pressure injection or osmotic pump |
Mook et al. 2007 |
| Green fluorescent protein, survivin | Glioma | FI | Nanoparticle-bound siRNA | Mouse, ectopic glioma xenograft | IV injection |
Medarova et al. 2007 |
| Green fluorescent protein | Melanoma | FI | siRNA | Mouse, B16F10-EGFP melanoma | Electropulsation |
Golzio et al. 2007 |
| cAMP-dependent protein kinase A | N/A | Gamma-camera | 99mTc-siRNA | ACNH human renal adenocarcinoma xenografts | IV injection |
Liu et al. 2007 |
| Ribonucleotide reductase M2 subunit | Cancer | BLI | siRNA | Mouse, transfected neuroblastoma xenograft | Pressure injection |
Heidel et al. 2007 |
| Ribonucleotide reductase M2 subunit | Cancer | BLI | SiRNA/CDadamantane-PEG nanoparticles non-targeted Tf-targeted | Mouse, neuroblastoma xenograft | IV injection |
Bartlett and Davis 2007a |
| Alpha-v integrin | Cancer | BLI | IVT siRNA, liposomal complex | Mouse PC3-luc prostate xenograft | Local, intratumoral injection |
Bisanz et al. 2005 |
| HIF-1a | Glioma | BLI | SiRNA-JetPEI complex | Mouse U251-HRE glioma HIF-1a/luc reporter | Local, intratumoral injection |
Gillespie et al. 2007 |
| CXCR4 | Breast cancer | BLI, MRI | shRNA | Mouse, orthotopic and disseminated cancer | IV injection |
Smith et al. 2004 |
| Keratin K6a mutation | Pachyonychia congenita | BLI | shRNA, bicistronic vector | Mouse, skin | Local, intradermal | |
| P-glycoprotein | Cancer | BLI | shRNA | Mouse, MDR1-Rluc MDR1-Fluc Fusion protein expression in tumor and liver cells | Pressure injection |
Pichler et al. 2005 |
- Luc, luciferase; BLI, bioluminescence imaging; FI, fluorescence imaging; PET, positron emission tomography; shRNA, small hairpin RNA; siRNA, small interfering RNA; IVT, in vitro transcription; IV, intravenous.
As noted above, one should use imaging marker genes for monitoring RNAi effects with caution. Firefly luciferase has a relatively short half-life in living cells (2–3 h) [Thompson et al., 1991]. In contrast, fluorescent proteins, which are otherwise excellent markers of gene expression have much longer half-lives (up to 26 h) [Mateus and Avery, 2000]. For example, in targeting of wild-type EGFR (epidermal growth factor receptor) or its deletion mutant in human glioma cell lines that were engineered to express a fusion between EGFR, Renilla luciferase and EGFP showed a correlation between the levels of receptor expression, cell proliferation and the luciferase signal [Arwert et al., 2007]. However, after knocking down the EGFR mRNA with the receptor-sequence specific shRNA vectors the observed cellular levels of EGFP did appear to correlate with the levels of EGFR protein, a fact that strongly favors the use of luciferases over other “imageable” expression marker proteins [Arwert et al., 2007].
However, there are cases when differential stability of lusiferase and EGFP expression in mammalian cells does not present a problem. For example, recently described delivery of siRNA therapeutics targeted the skin, one the most accessible sites for ‘organ-targeted’ therapy [Hickerson et al., 2007; Wang et al., 2007; Smith et al., 2008]. The authors injected into the skin a bicistronic expression vector with two marker cDNAs—EGFP and firefly Luc separated by a ribosome slippage site and positioned under the control of CMV promoter. The goal of the study initially was to test whether siRNA that was designed to target EGFP portion of mRNA was also resulting in a coordinated loss of luminescence due to a parallel knock-down of luciferase expression [Wang et al., 2007]. By co-injecting siRNA with the expression vector the authors demonstrated that luciferase bioluminescence (BLI) readout in vivo provided a sensitive way to observe RNAi sequence-specific effects that were directed at EGFP. The obtained in vivo imaging results suggested that by targeting a single mRNA encoding both the target sequence and the marker protein sequence, one could obtain an image reflecting the efficacy of siRNA. This approach permitted testing of siRNAs targeting keratin 6a mutation in a model of autosomal-dominant skin keratin disorder using similar co-injection of a bicistronic K6a/luciferase vector and siRNA [Hickerson et al., 2007; Smith et al., 2008].
Green fluorescence protein readout was also successfully used in a study where a multi-functional iron oxide nanoparticle was used as a vehicle for delivering covalently bound siRNA to tumors after the intravenous administration in vivo [Medarova et al., 2007]. Fluorescence imaging of bilateral ectopic glioma 9L tumors constitutively expressing either EGFP or red fluorescent protein (RFP) markers was performed 2 days after the single injection of nanoparticles that were covalently linked to siRNA duplexes. In addition, the nanoparticles were labeled with Cy5.5 dye (for visualization of nanoparticles), and linked to myristoylated polyarginine that was included to provide membrane translocating properties. Therefore, these particles could be imaged either by using MRI (superparamagnetic nanoparticles) or near-infrared imaging (due to Cy5.5 linking). While siRNA nanoparticle complex had no effect on RFP expression, EGFP expression was markedly decreased (Fig. 2C). Using intravenous delivery of survivin siRNA linked to the nanoparticles the authors reported 83% knock-down of survivin transcripts compared to a mismatch control RNA duplex. In view of MRI imaging results that showed focal (uneven) distribution of T2 contrast associated with particles in the tumor tissue, such extraordinary RNAi effect is clearly unexpected. Further details of high-efficacy nanoparticle-mediated siRNA delivery are currently under investigation [Medarova et al., 2007].
Luminescent readout is currently is the major approach to imaging of RNAi effects in live animals. Bioluminescent imaging was successfully used for detecting direct siRNA treatment effects in mouse model of prostate cancer. The siRNA was designed to down-regulate expression of human αV integrin subunit and were formulated with liposomal delivery vehicle [Bisanz et al., 2005]. The direct injection of siRNA complex in tumors reduced the growth of tumors implanted in the mouse tibia approximately fivefold if compared to liposomes only and had no effect on the tumors that were growing subcutaneously [Bisanz et al., 2005].
An indirect luminescence-based readout was used to track the RNAi effects targeted at hypoxia-induced factor-1 in U251 engineered glioma xenografts in vivo [Gillespie et al., 2007] (Fig. 2D). The authors assumed that if HIF1-alpha mRNA is targeted with siRNA, then, as a consequence, HRE (hypoxia responsive element)-controlled expression of firefly luciferase inevitably should respond to the decrease of HIF-1alpha levels. Indeed, after treating animals bearing ectopic tumors with multiple intratumoral injections of siRNA complexed with polyethyleneimine, the progressive decrease of HIF-1 mediated transactivation of HRE translated into measurable and statistically significant decrease of luminescence measured in tumors in live mice.
Luminescence imaging offers a semi-quantitative method of comparing the effectiveness of siRNA and shRNA expression vectors separately, as well as their combinations that usually result in an augmented RNAi effect [McAnuff et al., 2007] (Fig. 2E). If appropriate correction for tissue attenuation is implemented, luminescence light output can be easily converted into the expressed protein amounts. The authors observed that siRNA and shRNA vector were showing the same magnitude of knockdown, with a 10 µg dose achieving approximately 80% knockdown of the imaging signal measured in the liver. While siRNA and shRNA were nearly equipotent on a weight basis, shRNA was more efficient on a per mole basis in vivo. The authors speculated that the difference in RNAi effect was due to a faster degradation of siRNA in vivo and a higher affinity of endogenously transcribed siRNA to loading into RISC [McAnuff et al., 2007].
The further progress in developing in vivo imaging of RNAi depends on whether non-invasive and longitudinal approaches to tracking RNAi would be able to provide direct or indirect quantitative measures of target (as well as off-target) effects of RNAi. The ability to provide straightforward interpretation of the observed imaging effects is also extremely important. In some rare cases, for example, ferrochelatase present in colon cancer cells [Kemmner et al., 2007], RNAi effects are very easy to interpret since ferrochelatase knock-down translates in the increase of protoporphyrin-IX accumulation in cells which have resultant higher fluorescence emission in the range of 630–650 and 700 nm. This approach can be potentially translated into in vivo molecular imaging studies [Kemmner et al., 2007].
In conclusion, it is becoming apparent that detailed investigation of the mechanisms behind the RNAi effects exerted in vivo should include both imaging of distribution and silencing of relevant transcripts. The imaging of biodistribution would clearly benefit from better methods of stoichiometric labeling of RNA molecules with fluorine-18 and small chelates that can be used for tightly binding both positron and gamma-emitting metal cations. The techniques enabling imaging of gene knockdown would require quantitative and tomographic optical imaging approaches that are currently in development [Chaudhari et al., 2005; Zacharakis et al., 2005]. By combining of imaging of RNAi delivery (biodistribution) with the quantitative assessment of RNAi effects in vivo, researchers would have a better chance of transforming RNAi into a therapeutic modality.
Acknowledgements
The author is grateful to Michael Deleo (UMASS Medical School) for proofreading and helpful comments.

