

Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors
Abstract
Merkel cell polyomavirus (MCV) is a recently discovered human virus closely related to African green monkey lymphotropic polyomavirus. MCV DNA is integrated in ∼80% of Merkel cell carcinomas (MCC), a neuroendocrine skin cancer linked to lymphoid malignancies such as chronic lymphocytic leukemia (CLL). To assess MCV infection and its association with human diseases, we developed a monoclonal antibody that specifically recognizes endogenous and transfected MCV large T (LT) antigen. We show expression of MCV LT protein localized to nuclei of tumor cells from MCC having PCR quantified MCV genome at an average of 5.2 (range 0.8–14.3) T antigen DNA copies per cell. Expression of this putative viral oncoprotein in tumor cells provides the mechanistic underpinning supporting the notion that MCV causes a subset of MCC. In contrast, although 2.2% of 325 hematolymphoid malignancies surveyed also showed evidence for MCV infection by DNA PCR, none were positive at high viral copy numbers, and none of 173 lymphoid malignancies examined on tissue microarrays expressed MCV LT protein in tumor cells. As with some of the other human polyomaviruses, lymphocytes may serve as a tissue reservoir for MCV infection, but hematolymphoid malignancies associated with MCC are unlikely to be caused by MCV. © 2009 UICC
Merkel cell polyomavirus (MCV) DNA can be detected in 75–89% of MCC,1-4 an aggressive skin cancer of neuroendocrine origin.5, 6 MCV is a 5.4 kbp DNA virus that expresses tumor (T) antigen mRNAs in tumor tissues.7 The MCV large T (LT) gene sequences obtained from tumor-derived viruses have truncating mutations that are not present in wild-type virus. These mutations eliminate a helicase domain present in the C-terminus of the LT protein but preserve the N-terminal tumor suppressor retinoblastoma protein interaction site.7 Loss of viral helicase activity prevents MCV from actively replicating its own genome and thus the virus cannot be a passenger virus in tumors. A truncating mutation is also found in T antigen sequences from MKL-1, an MCC cell line harboring monoclonaly integrated MCV genome.8, 7 This cell line forms loose suspension aggregates in culture, the so-called classic phenotype.9 In contrast, MCC cell lines negative for the virus grow as tightly adherent, spindle-shaped cells (variant phenotype).7 Taken together, these findings strongly suggest that MCV causes a subset of MCC, a conclusion that would be strengthened by direct in situ detection of MCV oncoprotein expression in the MCC tumor cells.
Several primary and secondary malignancies are significantly associated with MCC, particularly chronic lymphocytic leukemia (CLL).10-13 It is not known if CLL or other lymphoid malignancies share a fundamental pathoetiologic link with MCC. MCV is most closely related to a lymphotropic polyomavirus (LPV) found in African green monkeys, the only polyomavirus known to naturally infect B lymphocytes.14 MCV has previously been found at low copy number in peripheral blood, as well as skin and gut tissues, of persons without MCC suggesting that it too may be lymphotropic.1 Systematic examination of peripheral blood for the presence of the virus, however, has not been previously described.
We show here that 70% of CK20 positive MCC tumors have uniform cancer cell expression of the MCV T antigen using a specific monoclonal antibody. Although MCV DNA is occasionally detectable at low levels in peripheral blood from MCC patients and healthy control donors, a survey of 325 hematolymphoid tumors failed to provide convincing evidence for high copy number MCV infection or evidence for T antigen expression in these tumors. This data suggests that MCV may be a common lymphotropic infection of humans as well as the cause for most MCC, but it is unlikely to directly contribute to hematolymphoid tumors that also occur in MCC patients.
Abbreviations
CLL, chronic lymphocytic leukemia; CK20, cytokeratin 20; GFP, green fluorescence protein; LPV, lymphotropic polyomavirus; LT, large T antigen; MCV, Merkel cell polyomavirus; MCC, Merkel cell carcinoma; PCR, polymerase chain reaction; PBMC, peripheral blood mononuclear cells; VP, viral protein.
Material and methods
Human tissue samples
For Merkel cell carcinoma, fresh frozen tumor samples were obtained from the Cooperative Human Tissue Network (CHTN). An MCC tissue core microarray consisting of 36 MCC specimens was generated from archival paraffin-embedded tissues from the pathology departments at Hospital Universitari del Mar and the Hospital Universitari Germans Trias i Pujol, Barcelona, Spain as previously described.15 Tissue microarrays for lymphoid malignancies and normal controls were purchased commercially (US Biomax, Inc.). Genomic DNA samples from consecutive hematolymphoid tumor tissues were collected and archived by the late Dr. Anne Matsushima, Columbia University, from excess tissue submitted for diagnostic pathology. This was supplemented with additional hematolymphoid tissues obtained from tissue banks at the Department of Pathology, University of Pittsburgh. For reasons of confidentiality, minimum patient identification and demographic data are available for most of these specimens. PBMC specimens were obtained from two sources: (1) excess samples submitted to the Division of Molecular Diagnostics, University of Pittsburgh Medical Center for genetic screening and (2) PBMC collected from HIV-positive persons participating in Kaposi's sarcoma epidemiologic studies.16 None of these study subjects were diagnosed with Merkel cell carcinoma. All specimens were tested under University of Pittsburgh Institutional Review Board-approved guidelines.
Real time quantitative PCR
Quantitative PCR was performed using primers amplifying the MCV T antigen, TAg (1051–1131 nt; forward: 5′-cctctgggtatggg tccttctca-3′, reverse: 5′-atggtgttcgggaggtatatc-3′) and VP2 (4563–4472 nt, forward: 5′-agtaccagaggaagaagccaatc-3′, reverse: 5′-ggc cttttatcaggagaggctatattaatt-3′) loci with internal TaqMan probes (TAg: 5′-cccaggcttcagactc-3′, VP2: 5′-gcagagttcctc-3′) labeled with FAM and MGB quencher (Applied Biosystems). For the additional 10 peripheral blood samples with CLL, primers designed against MCV T antigen promoter region (98–184 nt forward: 5′-cccaagggcgggaaactg-3′, reverse: 5′-gcagaaggagtttgca gaaacag-3′) and internal probe (5′-ccactccttagtgaggtagctcatttgc-3′) labeled with FAM and BHQ quencher (Biosearch Technologies) was used. Copy numbers were established from standard curves of Ct values from serial dilutions of known concentrations of MCV DNA originally amplified by PCR using contig 3 and contig 12 primer sets for TAg and VP2 detections, respectively.1 Water was used as control to detect template contamination. No evidence of PCR template contamination was observed in the PCR reactions with water control. RNaseP (Applied Biosystems) or β-actin primer-probe mixtures (forward: 5′-cactggctcgtgtgacaagg-3′, reverse: 5′-cagacctactgtgcgcctacttaa-3′, probe: 5′-tggtgtaaagcggccttggagtgtgt-3′) (Biosearch Technologies) were used to determine cell genome copy number. qPCR reactions were performed using PRISM 7700 Detection System, PRISM 7900HT Fast Real-Time PCR System (Applied Biosystems) and/or Smart Cycler 5RX4Z01 (Cepheid) with TaqMan reagents (UNG (+) TaqMan Universal PCR Master Mix). Amplification reactions of all target genes were performed with the following condition: 50°C for 2 min, denaturing at 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min. Results were expressed as numbers of viral copies per cell calculated from Ct values of viral and cellular gene standards (Table I). Cellular viral DNA copy number below 1.0 × 10−3 per cell was considered negative.
 |
Cell lines and transfection conditions
Human embryonic kidney 293 cells (ATCC) were grown in DMEM medium supplemented with 10% fetal calf serum. Cells were transfected with expression constructs using Lipofectamine 2000 (Invitrogen) following manufacturer's instructions on 90% confluent cells. Cells were harvested 48 hr after transfection for analysis.
Plasmids
To clone 57 kT wt, the genomic T antigen expression plasmid constructed from appendix 2067 was transfected into 293 cells with Fugene 6 (Roche). After 48 hr, total RNA was extracted with Trizol (Invitrogen). First strand cDNA was synthesized with oligo-d(T) primer using Superscript III First-Strand Synthesis System (Invitrogen). Multiply spliced products were amplified by PCR with primers: MCV.EcoRV(S) (5′-ccgatatcatggatttagtcctaaaagg-3′) and MCV.XhoI(AS) (5′-gggctcgagtattgagaaaaagtaccagaa tattggg-3′). A PCR fragment corresponding to 57 kT antigen was cloned into pcDNA6 expression vector with EcoRV and XhoI sites. To generate the pMCV TAg-EGFP expression constructs, pcDNA6 gLT206 encoding wild type full length genomic T antigen7 was digested with NheI and SacII and cloned into pEGFP-N1 (Clonetech) in-frame to C terminus GFP using same restriction sites. LT expression constructs for JCV and BKV were kindly provided by Dr. James DeCaprio.17 SV40 T antigen cDNA cloned in pCMV vector is described elsewhere.18
Generation of CM2B4 mAb
Monoclonal antibody CM2B4 (IgG2b isotype) was generated by standard methods of immunizing mice with KLH-derivatized SRSRKPSSNASRGA peptide from the MCV T antigen exon 2 with a C-terminal cysteine (Epitope Recognition Immunoreagent Core Facility, University of Alabama).
Immunofluorescence and immunohistochemistry
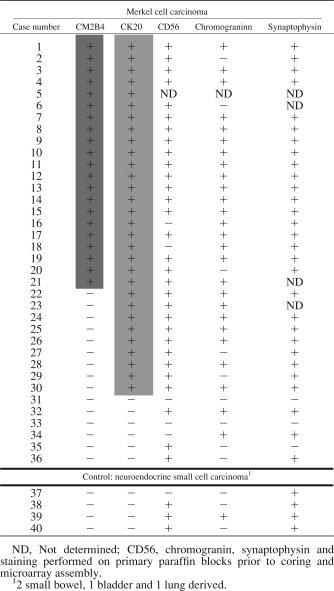
For immunofluorescence staining, cells were spotted on glass slides by Cytospin3 (Shandon), fixed with 10% buffered formalin for 20 min and permeabilized with phosphate-buffered saline (PBS) with 0.1% Triton X-100. After blocking, cells were reacted with CM2B4 (1:100 dilution) at 4°C overnight followed by secondary antibody (Alexa Fluor 488-conjugated anti-mouse, 1:1,000 Invitrogen) for one hour at room temperature. Stained cells were mounted in aqueous medium containing DAPI (Vector Laboratories, CA). For immunohistochemical staining of paraffin embedded tissues, epitope retrieval was performed using EDTA antigen retrieval buffer (Dako, Glostrup, Denmark) at 126°C for 3 min after deparaffinization and hydrogen peroxide treatment. After blocking with Protein Block (Dako), samples were reacted to primary antibody for 30 min at room temperature with dilutions described below. After washing, samples were incubated with Mouse Envision Polymer (Dako) for 30 min at room temperature for subsequent deaminobenzidine (DAB) reaction. mAbs used for immunohistochemistry were: CM2B4 (1:10–1:50 hybridoma supernatant), CK20 (Dako; 1:50), Chromogranin A (Dako, 1:600), Synaptophysin (Biogenex, San Ramón, CA; 1:100), and CD56 (Novocastra, Newcastle upon Tyne, United Kingdom; 1:50).
Immunoblotting
Transfected cells were lysed in buffer (10 mM Tris–HCl pH8.0, 0.6% SDS) containing proteinase inhibitor cocktail (Roche). Lysate was electrophoresed in 10% SDS-PAGE, transferred to nitrocellulose membrane (Amersham) and reacted with Pab416 (1:10) or CM2B4 mAb (1:10) for overnight at 4°C, followed by anti-mouse IgG-HRP conjugates (Amersham, 1:5,000) for 1 hr at room temperature. Detection of peroxidase activity was performed by Western Lightning plus-ECL reagent (Perkin Elmer).
Results
MCV and T antigen expression in Merkel cell carcinoma tumors
To show that MCC tumor cells are infected with MCV and express MCV T antigen protein, we developed a monoclonal antibody (CM2B4) to a peptide epitope (SRSRKPSSNASRGA) in exon 2 of the MCV T antigen. (Fig 1a). This epitope is located N-terminal to an LFCDE motif previously found to bind retinoblastoma protein and is likely to be conserved in viruses from both tumor and nontumor tissues.7 CM2B4 is predicted to target the MCV LT protein and a T antigen isoform translated from a multiply-spliced mRNA, but not the putative small T antigen (Fig. 1b). CM2B4 immunoblotting of lysates from 293 cells transfected with a wild-type genomic T antigen expression construct shows MCV LT protein migrating at ∼120 kDa and an additional ∼57kDa T antigen isoform translated from an alternatively spliced, SV40 17kT antigen-like mRNA (Fig. 1c). The identity of the 57 kDa protein was confirmed by transfection of a 57kT cDNA clone. T antigen loci cloned from tumor derived MCV sequences express faster migrating LT proteins with sizes corresponding to the presence of truncating mutations. The MCV infected MKL-1 cell line also expresses a truncated LT consistent with a premature stop codon mutation, whereas MCV negative UISO, MCC13, and MCC26 cell line lysates showed no specific reactivity (Fig. 1c).

(a) Amino acid alignment of MCV with known human polyomaviruses, SV40, and LPV. Region containing epitope site of CM2B4 is not conserved with these other polyomaviruses. (b) Transcript diagram from various wild-type and tumor-derived MCV T antigen loci. Yellow box represents epitope site and does not overlap with putative small T antigen (gray). (c) CM2B4 western blotting of constructs diagrammed in (b) shows detection of full length or truncated tumor LT antigens (open arrowheads) and 57kT antigen (solid arrowhead). Only the truncated LT antigen is seen in the MCV infected MKL-1 cell line, whereas no specific protein is detected from lysates of MCV negative UISO, MCC13 and MCC26 cell lines. Asterisk (*) denotes non-specific reactivity.
By immunofluoresence microscopy, precise nuclear colocalization of CM2B4 occurs with green fluorescence when an MCV LT-GFP fusion construct is expressed in 293 cells (Fig. 2a). CM2B4 is highly specific for MCV and does not react to T antigens from JCV or BKV by immunofluorescence (Fig. 2b) or to T antigens from JCV, BKV or SV40 by immunoblotting (Fig. 3a). In contrast, an anti-SV40 T antigen mAb, PAb416, cross-reacts with T antigens from other SV40-group viruses including JCV and BKV, but not with MCV T antigen (Figs. 2b and 3a). We examined 23 other anti-SV40 T antigen mAbs (Supporting Information TableI), and none show reactivity to MCV T antigen on immunoblotting. Tissue immunohistochemistry with CM2B4 of an MCV DNA positive MCC biopsy also shows strong reactivity among tumor cells, but not surrounding tissues (Fig. 2c). CK20, a low molecular weight cytokeratin marker for MCC19-22 has a characteristic perinuclear dot-like pattern in CM2B4 positive cells (Fig. 2c). Similarly, examination of the MCV positive, MCC-derived MKL-1 cell line shows expression of LT protein in a diffuse nuclear distribution (Fig. 3b). CM2B4 does not react to JCV T antigen in a brain section with progressive multifocal leukoencephalopathy (Fig. 3c).

(a) MCV T antigen-EGFP fusion protein (green) colocalizes with CM2B4 staining (red) in 293 cells transfected with pMCV TAg-EGFP or pEGFP, an empty EGFP vector. (b) Immunofluorescence staining of 293 cells over-expressing T antigen constructs from JC, BK and MCV shows that CM2B4 specifically detects MCV T antigen. PAb416 shows reactivity with JC and BK T antigens, but not with MCV T antigen. (c) Sequential sections from an MCC skin excision shows small, round blue sheets of tumors cells (H&E) which express MCV LT (CM2B4) and CK20 proteins. Expression of MCV LT protein shows a diffuse nuclear pattern in MCC tumor cells but not in surrounding tissues including the epidermis, adnexal epithelium (arrow), endothelial cells or dermal fibroblasts.

(a) Constructs encoding LT genes for JCV, BKV, SV40 and MCV were expressed in 293 cells and cell lysates were immunoblotted with CM2B4 or PAb416 antibodies. PAb416 cross-reacts with JCV and BKV LT proteins but not with MCV LT. (b) CM2B4 detects robust T antigen expression in a diffuse nuclear pattern in each cell from the MCV positive MKL-1 cell line. These cells retain perinuclear CK20 expression seen in Merkel cell carcinomas. (c) Brain tissue with progressive multifocal leukoenchephalopathy shows JCV infection of oligodendroglial cells by JCV specific in situ hybridization (left panel), and CM2B4 shows no reactivity to JCV antigens (right panel).
These results were extended by examining a tissue microarray containing 30 CK20-positive MCC, 6 CK20-negative but clinically suspect MCC, and 4 CK20-negative neuroendocrine control tumors. Of the 30 CK20 positive MCC, 21 (70%) were positive for LT protein expression. This suggests that the majority of MCC is caused by MCV infection whereas a subset of CK20+ MCC may have a different etiology. Of the 6 CK20-negative tumors diagnosed as MCC, none were positive for MCV LT protein expression but variably expressed neuroendocrine markers CD56, synaptophysin, or chromogranin (Table I).
To correlate MCV LT protein expression with the level of MCV infection, we next developed a quantitative real-time PCR (qPCR) assay. DNA from ten tumors previously examined1 were tested with primers designed to amplify regions of the T antigen and VP2. Seven of these Southern blot positive tumors had an average of 5.2 (range 0.8–14) T antigen DNA copies per cell consistent with clonal integration and viral genome concatenation (Table II). Similar results were found using VP2 qPCR except for MCC345 and MCC347. These tumors had robust T antigen qPCR positivity (5 copies per cell) but minimal amplification of the VP2 gene, which may reflect integration and loss of this late viral capsid gene region as has been described by Kassem et al.2 CM2B4 staining in CK20+ MCCs was concordant with qPCR results for all cases except MCC344, which showed abundant viral DNA but was negative with CM2B4 staining (Table II).
| MCC tissue | MCV genome copies per cell1 | Genomic Southern2 | Immunostaining | ||
|---|---|---|---|---|---|
| TAg | VP2 | CM2B4 | CK203 | ||
| MCC337 | <10−3 | 0 | − | − | + |
| MCC339 | 5.2 | 11.1 | + | + | + |
| MCC343 | 0 | 0 | − | − | + |
| MCC344 | 6.3 | 13.7 | + | − | + |
| MCC345 | 4.9 | <10−3 | + | + | + |
| MCC346 | <10−3 | 0 | − | − | + |
| MCC347 | 1.6 | 0 | + | + | + |
| MCC349 | 3.3 | 8.0 | + | + | + |
| MCC350 | 0.83 | 3.0 | + | NT4 | NT |
| MCC352 | 14.3 | 47.5 | + | + | + |
- 1 RNaseP copy number was divided by two to determine cellular equivalent of DNA.
- 2 MCV positivity was previously examined by Southern blotting (1).
- 3 CK20 expression was previously examined by immunostaining (1).
- 4 NT, no paraffin embedded MCC tissues to evaluate.
PBMC infection with MCV
To assess MCV lymphotropism as seen in other human polyomaviruses,23, 24 83 whole PBMC DNA samples collected from persons undergoing genetic testing for Factor V Leiden deficiency were tested by qPCR. These samples were collected mainly from adults (average of 60 years, range 1–78 years) with 73 (88%) samples from persons over 18 years of age. None were positive for viral DNA. However, among 21 PBMC collected from adult HIV/AIDS patients without MCC, 2 (9.5%) were positive by either T antigen (2.8 × 10−3 copies per cell) or VP2 (8.8 × 10−3 copies per cell) primers and one (5%) was positive with both primers (T antigen, 7.9 × 10−3 copies per cell; VP2, 6.0 × 10−3 copies per cell). Although we are unable to verify MCV lymphotropism in this study since examining whole PBMC is unlikely to reliably detect MCV due to the dilution effect from nonpermissive PBMC, increased rates of detection in HIV positive individuals suggest that immunosuppression may lead to reactivation of virus.
Survey of hematolymphoid malignancies for MCV infection
qPCR was further performed on tissue DNA from 104 T cell-associated and 161 B cell-associated malignancies, 19 myeloid disorders and 41 other tumors including Hodgkin lymphoma and post-transplant lymphoproliferative disorders (Table III). Of these 325 tumors, 7 (2.2%) were positive for either T antigen or VP2 DNA and two were positive for both. No consistent pattern of virus infection was found among these malignancies: 1 (3%) of 33 chronic lymphocytic leukemia, 1 (7.1%) of 14 non-Hodgkin lymphoma, not otherwise specified (NOS), 2 (3.1%) of 65 diffuse large B cell lymphoma, 1 (11%) of 9 marginal zone lymphoma and 1 (3.3%) of 30 Hodgkin lymphoma (Table III). Copy numbers for these positive hematolymphoid malignancies (Supporting Information Table II), however, were all 2–4 logs lower than MCV-positive MCC tumors (Table II). These results were confirmed by CM2B4 staining of commercial tissue microarrays of hematolymphoid tumors. Of 122 B cell lymphomas, 17 T cell lymphomas, one myeloid disorder and 2 Hodgkin lymphomas examined, none showed evidence for LT protein expression (Supporting Information Table III). Thirty-one healthy lymphoid control tissues were also negative for MCV T antigen.
| Hematopathological samples studied | No. tested | No. MCV positive (% MCV positivity) |
|---|---|---|
| B-cell-associated lymphomas | ||
| Chronic lymphocytic leukemia | 33 | 1 (3.0) |
| Non-Hodgkin lymphoma, NOS | 14 | 1 (7.1) |
| Diffuse large B cell lymphoma | 65 | 2 (3.1) |
| Follicular lymphoma | 14 | 0 |
| Acute lymphoblastic leukemia | 11 | 0 |
| Primary effusion lymphoma | 2 | 0 |
| Mucosa-associated lymphoid tissue lymphoma | 5 | 0 |
| Mantle cell lymphoma | 8 | 0 |
| Marginal zone lymphoma | 9 | 1 (11) |
| T-cell-associated lymphomas | ||
| Acute lymphoblastic leukemia | 10 | 0 |
| Large granular lymphocyte leukemia | 1 | 0 |
| Mycosis fungoides | 11 | 0 |
| T-cell lymphoma, unspecified | 82 | 1 (1.2) |
| Myeloid disorders | ||
| Chronic myelogenous leukemia | 5 | 0 |
| Acute myeloid leukemia | 11 | 0 |
| Myelodysplastic syndrome | 3 | 0 |
| Others | ||
| Hodgkin lymphoma | 30 | 1 (3.3) |
| Post-transplant lymphoproliferative disorder | 11 | 0 |
| Total | 325 | 7 (2.2) |
MCV and chronic lymphocytic leukemia
Given the epidemiological relationship between MCC and CLL, we examined additional CLL cases for evidence of MCV infection. Ten peripheral blood samples with CLL (WBC counts ranging from 13.2 × 109 − 84.3 × 109 cells per L) were harvested and tested for the presence of MCV DNA. One displayed low MCV positivity VP2 (2.0 × 10−3 copies per cell). Twelve additional paraffin-embedded biopsies with CLL were examined by CM2B4 staining only. All CLL cases were uniformly negative for MCV T antigen protein expression.
Discussion
Several independent groups have confirmed that MCV infection is present in the majority of MCC but not in most tissues from control patients.1-4 The monoclonal pattern of MCV infection1 and the pattern of tumor-virus derived mutations in T antigen genes7 provide strong evidence that MCV causes a subset of MCC. We now show that MCC tumor cells, but not adjacent non-neoplastic cells, are infected with MCV and express MCV T antigen protein in most MCV-positive MCC tumors. This provides additional evidence that MCV is a human tumor virus.
Quantitative PCR shows that MCC biopsies are generally positive for the virus at >1 copy per tumor cell. This is consistent with clonal integration being common in MCV-positive MCCs and with antibody staining results that revealed nearly all MCV-positive MCC tumor cells harbor virus. Expression of the MCV T antigen in tumor cells may provide clues on the molecular origins of these tumors since T antigens from other polyomaviruses have been found to be oncoproteins. Animal models show that continued expression of polyomaviral T antigen, for example, is required to induce and maintain viral transformation.25, 26 Additional studies are needed to determine whether MCV LT protein or other alternatively spliced T antigen isoforms contribute to cell transformation.7 Our study does not provide this formal proof but it does demonstrate that T antigen is a useful marker for MCC tumor cells. If MCV T antigen protein(s) are shown to play a direct role in MCC formation, they would be a unique nonhuman target for specific anticancer therapy.
Although a substantial fraction of adult humans may be infected with MCV,1 we only find evidence for high virus copy number infection in MCC. Approximately 70% CK20+ MCCs were positive for LT protein expression in our study. This suggests that the majority of MCC is caused by MCV infection whereas a subset of CK20+ MCC may have a different etiology. Other clinically-suspect MCCs lacking CK20 expression, were all negative for LT protein expression. Additional clinical validation is needed for CM2B4 staining as a tissue marker for MCV, but this preliminary evidence suggests it is useful in identifying subsets of tumors associated with MCV infection.
We find little support for the possibility that MCV also contributes to CLL, a malignancy that can be associated with MCC, or other common hematolymphoid malignancies. Methods in sample collection did not allow us to screen specimens simultaneously for both viral DNA and T antigen expression, but results from both assays were consistent and no CLL cases were MCV positive at levels found among MCCs infected with the virus. MCC arising in the setting of CLL may result from CLL-induced immunosuppression.13 These results do not exclude the possibility that less common hematolymphoid or nonlymphoid malignancies might be caused by this virus, and surveys of other tumors and diseased tissues are warranted.
Quantitative PCR detects MCV infection in peripheral blood cells from infected persons but at levels that approach the technical limit of our technique (∼10−3 DNA copies per cell). This is similar to other lymphotropic viruses, such as Kaposi's sarcoma herpesvirus, in which only a fraction of PBMC are positive from persons known to be infected with the virus.16, 27 Examining whole PBMC is unlikely to reliably detect MCV in most infected persons due to the dilution effect from nonpermissive peripheral blood cells. For this reason, it is not surprising that low rates of positivity are present for whole PBMC samples from persons screened for an unrelated genetic marker (Factor V Leiden) or HIV/AIDS patients. Determining true human prevalence for MCV infection will require development of serologic tests that can accurately measure exposure to this virus.28
Acknowledgements
We thank Dr. Mary Ann Accavitti for production of CM2B4; Ms. Marie Acquafondata and Ms. Yuan-Yuan Chen for tissue staining; Dr. Clayton Wiley for providing the PML case; Drs. Linda Gooding and James Pipas for SV40 T antigen antibodies; Dr. James DeCaprio for BKV and JCV T antigen constructs and Dr. Montserrat Gilaberte for reviewing the clinical histories of MCC tissue microarray patients. We thank Ms. Chrissie Usher and Ms. Susan Scudiere for data management. YC and PSM are funded as American Cancer Society Research Professors.

