

Clinical and genetic screening in a large Iranian family with Marfan syndrome: A case study
Farzane Vafaeie and Zahra Miri Karam contributed equally as the first authors.
Abstract
Background and Aims
Marfan syndrome (MFS) is an autosomal dominant genetic disorder caused by pathogenic variants of the fibrillin-1-encoding FBN1 gene that commonly affects the cardiovascular, skeletal, and ocular systems. This study aimed to evaluate the clinical features and genetic causes of the MFS phenotype in a large Iranian family.
Methods
Seventeen affected family members were examined clinically by cardiologists and ophthalmologists. The proband, a 48-year-old woman with obvious signs of MFS, her DNA sample subjected to whole-exome sequencing (WES). The candidate variant was validated by bidirectional sequencing of proband and other available family members. In silico analysis and molecular modeling were conducted to determine the pathogenic effects of the candidate variants.
Results
The most frequent cardiac complications are mitral valve prolapse and regurgitation. Ophthalmic examination revealed iridodonesis and ectopic lentis. A heterozygous missense variant (c.2179T>C/p.C727R) in exon 19 of FBN1 gene was identified and found to cosegregate with affected family members. Its pathogenicity has been predicted using several in silico predictive algorithms. Molecular docking analysis indicated that the variant might affect the binding affinity between FBN1 and LTBP1 proteins by impairing disulfide bond formation.
Conclusion
Our report expands the spectrum of the Marfan phenotype by providing details of its clinical manifestations and disease-associated molecular changes. It also highlights the value of WES in genetic diagnosis and contributes to genetic counseling in families with MFS.
1 INTRODUCTION
Marfan syndrome (MFS; MIM #154700) is an inherited connective tissue disorder with considerable clinical variability. It is mostly defined by a wide range of clinical signs affecting multiple systems and organs, including the skeletal, ocular, and cardiovascular systems, as well as the skin, lung, and dura. The primary cause of death in patients with MFS is aortic complications.1 It was first described in 1896 by Antoine-Bernard in a 5-year-old girl with skeletal problems.2 The incidence rate of MFS is approximately 0.5 to 1 in 5000 individuals in the general population, with no gender or ethnic bias.3 The clinical diagnosis of MFS is based on a combination of skeletal, ophthalmic, and cardiovascular symptoms and complications along with a positive family history, if available. In the absence of family history, genetic testing may be used to confirm the diagnosis. According to the revised Ghent criteria,4 several features, including dislocated lenses of the eye and aortic root aneurysm, are sufficient for the unmistakable diagnosis of MFS with or without a positive family history. In most cases, MFS is inherited in an autosomal dominant pattern and is caused by homozygous, heterozygous, and compound heterozygous mutations in the Fibrillin-1 (FBN1, OMIM ID: #134797, NCBI Gene ID: 2200) gene. To date, more than 3000 pathogenic variants of FBN1 have been reported in MFS in the Universal Mutation Database (http://www.umd.be). Most pathogenic variants are exclusive to each MFS family, while only 10%−15% of variants recur between different families.5 The FBN1 protein undergoes cysteine substitution via a missense mutation, which is the most common pathogenic variant. Other types of mutations inclue nonsense mutations, deletions, insertions, various types of splice site mutations, and rarely large deletions of FBN1.6 According to data from the Universal Mutation Database (UMD, http://www.umd.be/FBN1), more than 1500 pathogenic variants have been reported in the FBN1. The FBN1 gene contains 66 exons, located at the 15q21.1 chromosomal region, and encodes the FBN1 protein, a large extracellular matrix (ECM) glycoprotein (2871 aa, >350 kDa) that is, highly conserved among different species. One of the most reliable and enduring associations between genotype and phenotype occurs when the mutation disrupts the cysteine residue, resulting in ectopia lentis. MFS patients with mutations that cause premature termination of translation are more prone to skeletal abnormalities, while ectopia lentis is less common.7 Extracellular microfibrils, which are essential tissues components that endure repeated stretching and recoil, are primarily composed of FBN1. This protein is present in both elastic and nonelastic connective tissues throughout the body and is often closely associated with elastin fibers. FBN1 plays an important role in the stability and strength of these tissues.8 Microfibril production is disrupted by pathogenic FBN1 variants, which also cause abnormal fibrillin protein formation and ultimately impair connective tissue.9
Here, we characterized the phenotype and determined the genetic etiology of a large Iranian family with symptoms consistent with MFS using molecular diagnostic methods. Through whole exome sequencing (WES) and bioinformatics analysis, a heterozygous missense pathogenic variant (c.2179T>C) in FBN1 gene was detected in the proband and affected family members. Moreover, we performed molecular docking to evaluate the effect of this variant on protein−protein interactions.
2 MATERIALS AND METHODS
2.1 Family presentation
Our proband (II:2, indicated by an arrow in the pedigree, Figure 1) was a 46-year-old female patient with general skeletal, visual, and cardiovascular problems who was referred to the genetic counseling center in Birjand in April 2023. The demographic information, detailed clinical data, and family history of the proband and her family were carefully assembled. She was the second sibling of non-consanguineous parents, originally from eastern Iran. The onset of ocular problems occurred when she was 20 years old with low visual acuity and slow progressive vision loss. The proband had a height of 178 cm and body mass index of 17 kg/m2. She had a history of heart and brain strokes and was regularly taking warfarin. The son of the proband (III:4), 16 years old, had a Marfan phenotype. He had various clinical features including mitral valve prolapse, orthodontic problems, thumb and wrist signs, pulmonary blebs (pneumothorax), striae distensae, and pectus deformity. The wrist signs of the proband and her son are shown in Figure 2A,B, respectively. The proband's echocardiography findings are shown in Figure 3C. The pedigree chart was created using Evagene pedigree drawing software (https://www.evagene.com/). Other family members (II:6, III:13, III:14, IV:1, IV:2, IV:3, and IV:4) had a normal phenotype. The procedure was approved by the Medical Ethics Review Board of Birjand Medical University (no. IR.BUMS.REC.1394.468) and the investigation was conducted in compliance with the principles outlined in the Declaration of Helsinki. Clinical and genetic evaluations were approved by the proband and family members, and informed consent was obtained from each participant.



2.2 Clinical evaluations
The revised Ghent criteria were used for the proband and all suspected MFS-family members.10 Certified clinicians conducted a detailed clinical history, laboratory tests, and standard physical examinations. The affected family members underwent cardiovascular evaluations, including cardiac ultrasonography and CT scans of the entire aorta. Chest and finger X-rays to evaluate the skeletal system. Moreover, routine ophthalmic examinations were performed by an experienced ophthalmologist, including visual acuity measurements, slit-lamp biomicroscopy, retinoscopy, tonometry, and fundus examination.
2.3 Genetic evaluations
Peripheral blood samples from our affected proband, available family members (n = 14), and 10 unrelated subjects with normal family history and phenotype were collected for DNA extraction. DNA was extracted from the lymphocytes using the phenol/chloroform method described by Miller et al.11 The quality of the extracted genomic DNA was measured using an Epoch spectrophotometer (BioTek Inc.). To identify the genetic alterations causing this phenotype, we performed WES on the patient's (II:2) DNA sample. Exons were enriched, and the library was constructed using the Agilent SureSelect Human All Exon Version 6 Kit and TruSeq Exome Enrichment Kit. Libraries was sequenced using an Illumina NovaSeq. 6000 instrument (Illumina) according to the manufacturer's standard operating protocols. The short reads obtained were aligned to the UCSC hg19 reference genome using the NovoAlign software package. Picard Toolkit v.2.14.0 (http://picard.sourceforge.net), and the FastQC software12 (https://bioinformatics.babraham.ac.uk) were used to exclude duplicate reads and control the quality of reads, respectively. Sequence data conversion/indexing, variant determination, and functional annotation were performed using SAM (http://samtools.sourceforge.net), GATK v3.4.0 (Genomic Analysis Toolkit),13 and ANNOVAR14 toolkits. Rare variants with a minor allele frequency lower than 0.01 were screened using international online variant databases, including ExAC (http://exac.broadinstitute.org/), 1K Genome Project Phase 3 (http://www.internationalgenome.org/data), NHLBI ESP (Exome Sequencing Project, https://evs.gs.washington.edu/EVS), GME Variome (Greater Middle East, https://igm.ucsd.edu/gme/), and Iranome (A catalogue of genomic variations in the Iranian, http://iranome.ir/) databases. The frequencies of the selected variants were checked in the Iranome database (https://www.iranome.ir/), and their clinical significance was determined using the ClinVar (https://ncbi.nlm.nih.gov/clinvar/), OMIM (Online Mendelian Inheritance in Man, https://omim.org), and HGMD (Human Gene Mutation Database, http://hgmd.cf.ac.uk) databases. To verify suspected pathogenic variants and perform cosegregation analysis, polymerase chain reaction (PCR) and bidirectional sequencing were performed on the patient and her available family members on an ABI 3730xl DNA analyzer (Applied Biosystems). We conducted Sanger sequencing to confirm mutation in the proband (II:2) and her son (III:4), and also, we analyzed the mutation in DNA samples from other available family members (13 individuals including II:1, II:3, II:4, II:5, III:1. III:2, III:3, III:5, III:8, III:9, III:10, III:11, and III:12) by ARMS-PCR. PCR amplification of the target DNA region was performed under the following standardized PCR conditions: initial denaturation at 95°C for 5 min (during the holding cycle), followed by 30 cycles of denaturation at 95°C for 15 s, annealing at 51°C for 30 s, and extension at 72°C for 30 s. A standardized master mixture (30 µL) was prepared by combining 15 µL of Taq DNA Polymerase 2X Master Mix Red (Ampliqon), 12 µL of ddH2O, 1 µL of DNA template, and 1 µL of each forward and reverse primer (10 pM). Primer sequences were used for Sanger sequencing are summarized in Table 1. The sequencing results were analyzed using SnapGene Viewer v6.2.2 software (GSL BioTech LLC). Candidate variants and pathogenicity levels were determined according to ACMG/AMP standard guidelines.
| Gene | Primer sequence (5′→3′) | Primer length (bp) | Tm (°C) | Amplicon size (bp) |
|---|---|---|---|---|
| FBN1 (exon 19) | Forward: GTATTTATTTTATAATCTTAATTGATTTTGA | 31 | 53 | 200 |
| Reverse: TTCAGAAAATGGGTAAAACTTCT | 23 | 54 |
- Abbreviation: PCR, polymerase chain reaction.
2.4 Bioinformatic evaluations
To assess the possible effect of the identified variants on protein function, candidate variants were analyzed using several computational predictors, including PolyPhen-2 (Polymorphism Phenotyping version 2, http://genetics.bwh.harvard.edu/pph2/), MutPred (http://mutpred.mutdb.org/), FATHMM (http://fathmm.biocompute.org.uk), SIFT (Scale Invariant Feature Transform, http://sift.jcvi.org/), EIGEN, LRT (Likelihood Ratio Test), PROVEAN (Protein Variation Effect Analyzer, http://provean.jcvi.org/seq_submit.php), and MutationTaster2 (http://mutationtaster.org). We used the Combined Annotation-Dependent Depletion (CADD) tool, available at https://cadd.gs.washington.edu, to evaluate the pathogenicity scores of the identified variants. Moreover, alignment of the FBN1 protein sequence among different species (human, mouse, Norway rat, cattle, chicken, rhesus, zebrafish, etc.) was performed using the EMBL-EBL Kalign tool15 (https://www.ebi.ac.uk/Tools/msa/kalign/). Phylogenetic comparisons were performed to examine vertebrate conservation using PhastCons100 (http://compgen.bscb.cornell.edu/phast/) and the PhyloP100 conservation tools. STRING Database v11.5 (https://string-db.org/cgi/network) was used to investigate for the physical protein−protein interactions of FBN1 with other existing proteins.
2.5 Molecular docking
Using the Iterative Threading Assembly Refinement (I-TASSER) server, we conducted ab initio/threading-based three-dimensional structure predictions for wild-type and mutant FBN1 proteins. The quality of the models was evaluated based on the confidence score (C-score), which ranged from −5 to 2 and were considered acceptable. The RAMPAGE server was used to evaluate the secondary structure of the models. To determine the precise affinity between FBN1 (wild-type and mutant) and latent transforming growth factor beta (TGF-β) binding protein 1 (LTBP1), we performed molecular docking and calculated the interaction energy using the HEX server. The PyMOL molecular graphics system was used to visualize possible interactions between the structural models. Sanger sequencing was performed to validate the newly identified potential causative variants.
3 RESULTS
3.1 Clinical findings
A total of 17 related patients with a definite or suspected clinical diagnosis of MFS were recruited for the study. The patients included 11 males and six females, from one family in the South Khorasan Province (Figure 1). Physical, ophthalmic, and cardiovascular examinations were performed by an ophthalmologist and a cardiologist (Table 2). Patients present with a range of intricate symptoms, including cardiovascular, ocular, and skeletal abnormalities. All of these patients in the family manifested various visual problems, including iridodonesis (17/17), high intraocular pressure with ectopia lentis (17/17), cataracts (1/17), and blindness (3/17). Mitral valve prolapse and regurgitation followed by aortic root dilation were observed in 16 of the 17 affected individuals. The general physical features of the patients included tall stature, arachnodactyly, disproportionately long and thin limbs, and joint laxity. The wrist signs in the proband and her son are shown in Figure 2A,B, respectively. The proband's echocardiogram results are shown in Figure 3C.
| Case ID | Age (year) | Gender | Cardiac disease | Skeletal abnormalities | Ocular abnormalities |
|---|---|---|---|---|---|
| I1 | 67 (deceased) | Male | + | + | + |
| II1 | 49 | Female | + | + | + |
| II2 | 46 | Female | + | + | + |
| II3 | 43 | Male | + | + | + |
| II4 | 41 | Male | + | + | + |
| II5 | 35 | Male | + | + | + |
| II6 | 33 | Male | − | − | − |
| III1 | 27 | Female | + | + | + |
| III2 | 22 | Female | + | + | + |
| III3 | 14 | Male | + | + | + |
| III4 | 16 | Male | + | + | + |
| III5 | 24 | Female | + | + | + |
| III6 | 13 (deceased) | Female | − | + | + |
| III7 | 7 (deceased) | Male | − | − | − |
| III8 | 18 | Male | + | + | + |
| III9 | 15 | Male | + | + | + |
| III10 | 14 | Male | + | + | + |
| III11 | 10 | Male | + | + | + |
| III12 | 8 | Male | + | + | + |
| III13 | 5 | Male | − | − | − |
| III14 | 1 | Female | − | − | − |
| IV1 | 4 | Female | − | − | − |
| IV2 | 3 | Male | − | − | − |
| IV3 | 1 | Female (identical twins) |
− | − | − |
3.2 Genetic and bioinformatics findings
Using next-generation sequencing, we identified a novel missense variant (c.2179T>C, p.C727R, cDNA.2635T>C, g.148470T>C) in exon 19 of FBN1 (GenBank NM_000138.5). The transcript contains 66 exons and is annotated with 438 domains and features. This substitution indicates that TGT (Cys) changes to CGT (Arg) at codon 727 located on exon 19 in EGF-like 11, which is a calcium-binding domain and has been suggested as a disorder variant. The proband was heterozygous for this variant. The FBN1 c.2179T>C variant has not been previously reported in the GME, ExAC, 1K Genome Project Phase 3, dbSNP, and Iranome databases. No previous description of the clinical significance of this variant has been found in clinical/genetic databases such as the Marfan database, ClinVar, HGMD, and OMIM. No other suspected or known variants with clinical significance were identified among the candidate variants. Subsequently, Sanger sequencing confirmed the mutation in the proband (II:2) and her affected son (III:4). Moreover, the presence of the mutation was determined by the ARMS-PCR method in other available family members (II:1, II:3, II:4, II:5, III:1. III:2, III:3, III:5, III:8, III:9, III:10, III:11, and III:12). They were also heterozygous, whereas no mutation was identified in his family members with normal phenotype. The sequencing results of the proband (exon 19 of FBN1) and her affected son are shown in Figure 3A,B, respectively. Based on the clinical data and genetic analysis, this missense variant was considered pathogenic. In silico analysis using several common web-based tools, such as PolyPhen-2, SIFT, PROVEAN, EIGEN, FATHMM, and Mutation Taster, predicted the p.C727R variant in FBN1 with pathogenic effects and altered protein function or structure. Using the CADD tool (a web-based tool for scoring the deleteriousness of variants from 1 to 99), the c.2179T>C variant was predicted to be a pathogenic variant with a score of 34. A summary of the results obtained from the in silico investigation is presented in Table 3. Multiple alignments among different species revealed that cysteine amino acid 727 of FBN1 is evolutionarily conserved in all investigated vertebrates (Figure 3C) and plays an important role in protein function. To detect the possible interactions between the FBN1 protein and other existing proteins implicated in MFS pathogenesis, we searched the STRING database, as illustrated in Figure 4, and predicted a physical protein−protein interaction network. At an interaction score of 0.900, which represents the highest confidence level, the results revealed that the FBN1 protein physically interacts with six other proteins. These proteins included LTBP1, ADAMTS10, EL1, FN1, FBLN2, and MFAB2. Understanding these interactions is essential to elucidate the molecular mechanisms underlying MFS and to help select candidate proteins for molecular docking studies with FBN1.
| Algorithm | Prediction | Score |
|---|---|---|
| PhyloP100 | Highly conserved | 8.94 |
| PhastCons100 | Highly conserved | 1.0 |
| SIFT | Damaging | 0.001 |
| Polyphen-2 | Probably damaging | 0.997 |
| LRT | Deleterious | 0 |
| PROVEAN | Damaging | −10.93 |
| PrimateAl | Pathogenic | 0.917 |
| Mutation Taster | Disease causing | 1.0 |
| MutPred | Pathogenic | 0.996 |
| FATHMM | Damaging | −5.91 |
| EIGEN | Pathogenic | 1.082 |
| M-CAP | Damaging | 0.969 |
| CADD | Deleterious | 27 |
| BayesDel addAF | Damaging | 0.577 |
| MetalR | Damaging | 0.990 |
| MetaSVM | Damaging | 0.967 |

3.3 Homology model building and molecular docking study
To better understand the molecular mechanics and structural consequences of the p.C727R mutation in FBN1, the online I-TASSER server was used to generate models of the EGF-like 11 domain, calcium-binding domain of Fibrillin, and C-terminal LTBP1 fragment for the mutant and wild-type. Table 4 presents the accuracy estimation and Ramachandran plots of the wild-type and mutant models of the calcium-binding domain and the C-terminal LTBP1 fragment. Ramachandran plots indicated that the majority of residues were in the favored region, with only a small number of outliers. To investigate the impact of the mutation on FBN1 binding, we conducted molecular docking studies using the Hex program (version 8.0.0). Figure 5 illustrates the docking results of FBN1 (wild-type and mutant) and LTBP1. Docking analysis revealed that substituting cysteine with arginine in the fibrillin led to a reduction of 23 kcal/mol. Furthermore, the results indicated that the binding pattern of the mutated Fibrillin was significantly distinct from that of the wild-type.
| Protein name | C-score | TM-score (mean ± SD) | RMSD (Å) | Ramachandran analysis | |||
|---|---|---|---|---|---|---|---|
| Plots | Favored | Allowed | Outlier | ||||
| FBN1-(wild) | 0.37 | 0.76 ± 0.10 | 1.7 ± 1.5 | 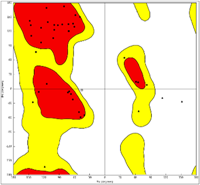 |
33 (78.5%) | 6 (14.4%) | 3 (7.1%) |
| FBN1-(mutant) | 0.30 | 0.75 ± 0.10 | 1.8 ± 1.5 | 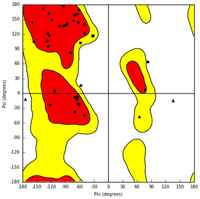 |
35 (83.3%) | 3 (7.2%) | 4 (9.5%) |
| LTBP1 | 0.91 | 0.84 ± 0.08 | 1.2 ± 1.2 | 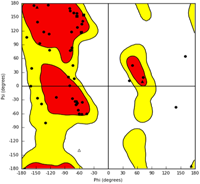 |
42 (76.4%) | 9 (16.3%) | 4 (7.3%) |
- Abbreviation: C-score, confidence score.
- The C-score serves as a confidence metric for evaluating the quality of predicted models generated by I-TASSER. Its computation relies on the assessment of threading template alignments' significance and the convergence parameters derived from structure assembly simulations. The C-score is typically reported within the range of −5 to 2, whereby a higher C-score indicates a model with greater confidence, while a lower C-score suggests lower confidence in the model's accuracy.
- TM-score is widely used for assessing structural similarity between protein structures, especially when the native structure is known. In our study, we utilized the C-score to estimate the TM-score of predicted models compared to native structures. This allowed us to determine the similarity or dissimilarity between the predicted models and native structures, providing insights into the accuracy of the modeling predictions. A TM-score >0.5 indicates a model of correct topology and a TM-score <0.17 means a random similarity. These cutoff does not depend on the protein length.

4 DISCUSSION
MFS is a genetic disorder with high morbidity that affects connective tissue. It is caused by pathogenic variants of FBN1 which can lead to serious life-threatening complications.16 Due to the significant variability of MFS phenotypic expression within and between families, delayed clinical manifestation in older ages, the high rate of spontaneous mutations, and presentation similar to other connective tissue diseases, diagnosing the condition can often prove challenging.17 In this study, we evaluated the clinical features and genetic causes of MFS in a large Iranian family. Moreover, we used molecular docking to investigate the interaction between the mutated FBN1 protein and LTBP1 protein to understand how mutations can affect the binding interface and potentially disrupt the interaction between the two proteins.
The proband was a 48-year-old woman with physical, cardiac, and ocular abnormalities. The patient was phenotypically similar to other patients with MFS. She was the second daughter of a nonconsanguineous parent. Her older sister also had the same clinical symptoms, but was more severe. We found that family members exhibited a wide variety of clinical features, including skeletal, cardiovascular, and ocular abnormalities. The clinical variability of MFS is remarkable, even among members of the same family, with the first symptoms appearing as well as the extent and severity of clinical presentations.18 Our findings were consistent with those of previous studies on MFS and emphasized the multisystemic nature of the disorder.
By evaluating all MFS patients in this large family, we found that mitral valve prolapse, regurgitation, and aortic root dilatation were the most common cardiovascular problems. Enlargement of the aortic root and proximal ascending aorta are the predominant cardiovascular problems observed in 80% of patients with MFS. This dilation can lead to aortic dissection, which is the primary cause of untimely mortality in these patients.19, 20
Among the most prominent clinical findings in this cohort, the patients had cardiovascular issues with mitral valve prolapse and regurgitation. Additionally, aortic root dilation was present in the majority of affected individuals. The prevalence of aortic root enlargement, which can ultimately lead to aortic dissection, is consistent with previous reports, and highlights the importance of cardiovascular monitoring and intervention in patients with MFS. Early detection and management of aortic root dilation are pivotal for reducing the risk of life-threatening complications associated with MFS.
On the other hand, a remarkable finding in the ocular system of the family was ectopia lentis (17/17) and iridodonesis (17/17). Ectopia lentis is a hallmark ocular feature of MFS, which refers to the displacement or dislocation of the eye's natural lens from its normal position in the eye. Many studies have reported that myopia and ectopia lentis are the most prevalent ocular signs of MFS, observed in over 60% of the cases.21, 22 Other notable observations include early and severe myopia, retinal detachment, cataracts, glaucoma, flat cornea, and iris hypoplasia.22 The high prevalence of ocular manifestations in our patients was consistent with well-established patterns of MFS, underscoring the necessity for regular ophthalmic evaluations in individuals with suspected or confirmed MFS.
We identified a missense pathogenic variant (c.2179T>C/p.C727R) in the FBN1 gene of the proband and 16 other family members by genetic analysis. All the patients were heterozygous for the FBN1 c.2179T>C variant. Genetic diagnosis serves as a crucial addendum to the proband's differential diagnosis, enabling a distinction between MFS and other syndromes, such as Shprintzen−Goldberg syndrome, Loeys−Dietz syndrome, and vascular Ehlers−Danlos syndrome. Although they have similar clinical features, these conditions arise from mutations in various genes. This study also highlights the value of WES in genetic diagnosis and as a contribution to genetic counseling in families with MFS. WES has become a widely used laboratory test for diagnosing MFS, owing to its superior resolution and accuracy at the whole-genome level.23, 24 Identifying the genetic cause of MFS is crucial for accurate diagnosis, prognosis, and management of the disease. The use of WES has allowed for a more comprehensive evaluation of the genetic cause of the condition, which is especially important in cases where clinical diagnosis is not straightforward.
FBN1 is a large ECM protein that plays an important role in the formation and maintenance of elastic fibers. Elastic fibers are a primary component of connective tissues such as the skin, blood vessels, and lungs and provide elasticity and resilience to these tissues.25 FBN1 interacts with a variety of proteins (Figure 4) to regulate the assembly and organization of elastic and fibrillin fibers within the ECM.26 One of the most well-known direct protein interactions of FBN1 is the glycoprotein LTBP1.26 LTBP1 is a chaperone protein that binds and sequesters latent TGF-β, which regulates cell proliferation and differentiation.27 The interaction between FBN1 and LTBP1 is complex and multifaceted. This interaction is important for the correct localization and assembly of LTBP1 in the ECM.28 FBN1 interacts with LTBP1 to target TGF-β in the ECM and facilitate its activation. Disruption of LTBP1 function can lead to various connective tissue disorders.29 Using molecular docking analysis, we found that the c.2179T>C/p.C727R variant located in the cbEGF domain of fibrillin, disrupts the interaction between FBN1 and LTBP1. Fibrillin contains EGF-like and cbEGF domains, which are responsible for binding to LTBP1. On the other hand, LTBP1 possesses an N-terminal domain followed by tandem repeats known as the TB domain, which interacts with fibrillin. The c.2179T>C/p.C727R variant of FBN1 is a missense mutation that alters a single nucleotide in the DNA sequence and leads to substitution of the amino acid cysteine with arginine at position 727 in the FBN1 protein. This mutation is located at the C-terminal of the FBN1 protein, which is important for its interaction with LTBP1. This variant may potentially affect the formation of disulfide bonds in the calcium-binding domain of the EGF-like 11 protein, leading to a significant functional alteration in the affected domain and its adjacent domains. This mutation reduces the binding affinity between FBN1 and LTBP1, resulting in impaired TGF-β activation and signaling. This may contribute to the pathogenesis of MFS, as dysregulation of TGF-β signaling is a key feature of this disorder. Overall, the use of molecular docking to investigate the interaction between FBN1 and LTBP1 can provide valuable insights into the molecular mechanisms underlying their complex relationship and can help to identify potential targets for therapeutic intervention in connective tissue disorders.18, 25, 26, 30, 31
5 CONCLUSION
In summary, we described a non-consanguinity MFS family with 17 affected members suffering from a rare pathogenic variant (c.2179T>C/p.C727R) in exon 19 of FBN1. Affected patients have various clinical features, including cardiovascular, ocular, and skeletal abnormalities. Clinical and genetic findings were consistent with a diagnosis of MFS, and in silico analysis predicted the pathogenic effects of the variant, suggesting alterations in FBN1 protein structure and function. These findings expand our knowledge of the genetic basis of MFS and highlight the significance of molecular genetic testing in the diagnosis and management of MFS. Additional investigations, especially functional studies, are warranted to validate the pathogenicity of the c.2179T>C/p.C727R mutation and elucidation of its precise molecular mechanisms in MFS. These results may have implications for clinical practice, potentially aiding in the diagnosis and management of MFS in affected individuals and their families. Molecular docking studies have suggested at altered binding patterns resulting from mutation. Further exploration of these structural changes could deepen our understanding of MFS at the molecular level. This research may pave the way for the development of targeted therapies or diagnostic tools for MFS, offering hope to affected individuals.
AUTHOR CONTRIBUTIONS
Farzane Vafaeie: Data curation; software; writing—original draft. Zahra Miri Karam: Investigation; methodology; software; writing—original draft. Abolfazl Yari: Formal analysis; investigation; methodology; software. Hossein Safarpour: Formal analysis; investigation; validation. Tooba Kazemi: Data curation; formal analysis. Shokoofeh Etesam: Conceptualization; data curation; resources. Mojtaba Mohammadpour: Data curation; methodology; resources. Ebrahim Miri-Moghaddam: Project administration; supervision; writing—review and editing. All authors have read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We would like to thank the family of the patient for their participation and consent for data publication in this study.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
This study was conducted in a manner compliant with the tenets of the Declaration of Helsinki. The Ethics Committee of Birjand Medical University gave its permission to our investigation (Ethics code: IR.BUMS.REC.1394.468). Written informed consent was obtained from all individual participants included in the study. All family members signed informed consent regarding publishing the clinical data.
TRANSPARENCY STATEMENT
The lead author Ebrahim Miri-Moghaddam affirms that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Open Research
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data, especially some clinical data, and patient images, are not publicly available due to privacy or ethical restrictions. The corresponding author (Ebrahim Miri-Moghaddam) had full access to all of the data in this study and took complete responsibility for the integrity of the data and the accuracy of the data analysis.

