

G-protein genomic association with normal variation in gray matter density
Abstract
While detecting genetic variations underlying brain structures helps reveal mechanisms of neural disorders, high data dimensionality poses a major challenge for imaging genomic association studies. In this work, we present the application of a recently proposed approach, parallel independent component analysis with reference (pICA-R), to investigate genomic factors potentially regulating gray matter variation in a healthy population. This approach simultaneously assesses many variables for an aggregate effect and helps to elicit particular features in the data. We applied pICA-R to analyze gray matter density (GMD) images (274,131 voxels) in conjunction with single nucleotide polymorphism (SNP) data (666,019 markers) collected from 1,256 healthy individuals of the Brain Imaging Genetics (BIG) study. Guided by a genetic reference derived from the gene GNA14, pICA-R identified a significant SNP-GMD association (r = −0.16, P = 2.34 × 10−8), implying that subjects with specific genotypes have lower localized GMD. The identified components were then projected to an independent dataset from the Mind Clinical Imaging Consortium (MCIC) including 89 healthy individuals, and the obtained loadings again yielded a significant SNP-GMD association (r = −0.25, P = 0.02). The imaging component reflected GMD variations in frontal, precuneus, and cingulate regions. The SNP component was enriched in genes with neuronal functions, including synaptic plasticity, axon guidance, molecular signal transduction via PKA and CREB, highlighting the GRM1, PRKCH, GNA12, and CAMK2B genes. Collectively, our findings suggest that GNA12 and GNA14 play a key role in the genetic architecture underlying normal GMD variation in frontal and parietal regions. Hum Brain Mapp 36:4272–4286, 2015. © 2015 Wiley Periodicals, Inc.
INTRODUCTION
Studying associations between genetic variables and imaging traits is a valuable strategy, a maturing field known as imaging genetics, which holds the promise to help better understand the genetic underpinnings of cognition and reveal biological mechanisms of mental disorders [Thompson et al., 2014]. Structural magnetic resonance imaging (sMRI) provides a noninvasive approach to study the morphology of the living brain. Quantitative measures are derived from T1-weighted MRI images using computational methods such as voxel-based morphometry (VBM) [Ashburner and Friston, 2005] or cortical surface reconstruction (FreeSurfer) [Fischl and Dale, 2000] to depict various structural features, including brain volume, gray matter volume, cortical thickness, and surface area. These features can then be compared at subject level for their functional implications. Under this strategy, biomarkers have been consistently identified from both healthy and diseased human brains, characterizing neural development, ageing, Alzheimer's disease, schizophrenia, and so on [Ellison-Wright et al., 2008; Frisoni et al., 2010; Giedd and Rapoport, 2010; Raz and Rodrigue, 2006]. More importantly, many attributes of brain structure, including both global and regional traits, are confirmed to be genetically influenced in twin studies, with heritability estimated around 40–90% [Peper et al., 2007; Thompson et al., 2001; Winkler et al., 2010].
Heterotrimeric guanine nucleotide-binding proteins (G-proteins) serve as molecular switches in intracellular singling cascades, where they sense signals from cell-surface receptors that are activated by extracellular stimuli and transduce signals to downstream effectors [Oldham and Hamm, 2008]. It has been documented that activated G-proteins directly interact with a variety of effector proteins, including phosphodiesterase E, phospholipase D, phospholipase C, inducible nitric oxide synthase, calcium channels, and the G protein-regulated inducer of neurite outgrowth 1 and 2 [Cabrera-Vera et al., 2003]. Particularly, G-proteins are richly expressed in the brain and involved in cortical development, neuronal growth as well as neuronal signaling [Bromberg et al., 2008; Hamm, 1998; Offermanns, 2001]. For instance, there is evidence that G protein alpha 12 and alpha 13 can mediate growth cone collapse and neurite retraction [Nurnberg et al., 2008], while deficiency of the G-protein α-subunits causes localized overmigration of neurons in the developing cerebral and cerebellar cortices [Moers et al., 2008]. Knockout of G protein beta 5 impairs brain development and causes multiple neurologic abnormalities in mice [Zhang et al., 2011]. Neural expression of G protein-coupled receptor 3, 6, and 12 has been implicated in up-regulating cyclic AMP (cAMP) levels in neurons and stimulating neurite outgrowth [Tanaka et al., 2007].
Given their roles in neural development, G-proteins pose promising candidates for imaging genetic association studies. More recently, Chavarria-Siles et al. [2013] investigated 502 single nucleotide polymorphisms (SNPs) in 25 G-protein genes for their associations with brain-wide gray matter volume using a mass univariate model. They identified seven SNPs to be significantly associated with gray matter volume variation in different brain regions, including the medial frontal cortex. While this work provides direct evidence that G-protein SNPs are related to brain structure, the univariate analysis is not able to assess the aggregate effects of multiple variants. In addition, considering that G-proteins are key cell signaling molecules and affect multiple biological processes, their effect on neurobiological conditions is likely not isolated but involves a large network. Therefore, a multivariate study is strongly encouraged to search for G-protein involved genetic components that may better delineate the basis of the gray matter variation.
In the present work, we applied a semi-blind multivariate approach, parallel independent component analysis with reference (pICA-R) [Chen et al., 2013], to explore G-protein involved genomic factors underlying brain structure in a large homogeneous cohort of 1,256 healthy Caucasians. Brain-wide gray matter density (GMD) images were analyzed in conjunction with genome-wide SNP data. G-proteins identified in the work by Chavarria-Siles et al. [2013] served as references to guide the analysis but the method still allows for other genetic variants to be revealed as well.
MATERIALS AND METHODS
Participants
BIG
A large cohort was used for discovery to increase the statistical power. This step was performed using the Brain Imaging Genetics (BIG) dataset, an ongoing effort being conducted at the Radboud University Nijmegen together with the Max Planck Institute for Psycholinguistics (Nijmegen, the Netherlands) [Bralten et al., 2011; Cousijn et al., 2012]. The regional medical ethics committee approved the study and all subjects provided written informed consent. Specifically in this work, a total of 1,256 healthy Caucasians were admitted into the investigation, including 617 males (age: 23.28 ± 4.11 years) and 843 females (age: 22.67 ± 3.61 years) for which both neuroimaging and genotype data were collected [Guadalupe et al., 2014]. All subjects are typically highly educated and free of neurological or psychiatric history according to self-report. To be noted, an overlap of subjects might exist between the present study and Chavarria-Siles et al.'s study. The latter employed 532 healthy BIG subjects.
MCIC
In the validation step, we used the subjects from the Mind Clinical Imaging Consortium (MCIC) study [Gollub et al., 2013], a collaborative effort of four research teams from University of New Mexico-Mind Research Network, Massachusetts General Hospital, University of Minnesota, and University of Iowa. The institutional review board at each site approved the study and all subjects provided written informed consent. Out of 255 subjects, 89 were healthy Caucasians, including 51 males (age: 31.41 ± 10.39 years) and 38 females (age: 33.61 ± 11.50 years). These subjects were employed to validate the results obtained from the BIG data. All healthy subjects were screened to ensure that they were free of any medical, neurological, or psychiatric illnesses, including any history of substance abuse.
Neuroimaging
BIG
Structural images were acquired at the Donders Centre for Cognitive Neuroimaging (Nijmegen, The Netherlands) using different scanners, i.e., 1.5 T Siemens Avanto and Sonata, as well as 3.0 T Siemens Trio and TIM Trio. Transmitting and receiving coils also differed across subjects. A standard sagittal T1-weighted three-dimensional magnetization prepared rapid gradient echo (MP-RAGE) sequence was employed, while some variations were observed in repetition, inversion, and echo time, as well as pixel bandwidth and flip angle. The use of parallel imaging with an acceleration factor of 2 was also included. Table 1 summarizes the settings used in BIG sMRI scans.
| Scanning parameter | Variations across subjects |
|---|---|
| Station name | avanto (462), sonata (160), trio (52), triotim (582), |
| Sequence name | *tfl3d1 (13), *tfl3d1_ns (983), spc3d1rr282ns (5), tfl3d1 (1), tfl3d1_ns (254), |
| Repetition time | 1660 (3), 1960 (13), 2250 (539), 2300 (615), 2730 (81), 3200 (5), |
| Echo time | 2.02 (3), 2.86 (1), 2.92 (22), 2.94 (1), 2.95 (462), 2.96 (183), 2.99 (14), 3.03 (348), 3.04 (1), 3.08 (1), 3.11 (1), 3.13 (1), 3.55 (1), 3.68 (148), 3.93 (51), 4.43 (7), 4.58 (3), 401 (5), 5.59 (3), |
| Inversion time | 1000 (81), 1100 (627), 750 (3), 850 (539), 900 (1), null (5), |
| Magnetic field strength | 1.494 (101), 1.5 (521), 2.89362 (52), 3 (582), |
| Number of phase encoding steps | 176 (3), 196 (5), 253 (5), 255 (565), 256 (677), 320 (1), |
| Pixel bandwidth | 130 (636), 140 (611), 240 (1), 260 (3), 751 (5), |
| Transmitting coil | body (1068), cp_head (49), txrx_head (139), |
| Flip angle | 120 (5), 15 (539), 7 (81), 8 (630), 9 (1), |
| Tcoil ID/receiving coil | 32ch_head (215), 8ch_head (573), body (1), cp_headarray (256), headmatrix (70), null (2), txrx_head (139), |
- Each scanning setting is followed by the number of subjects that have been scanned using this setting.
MCIC
The MCIC structural images were coronal T1-weighted MRIs collected at multiple sites. Table 2 lists the setting used in the scans. It can be seen that scanners differed among 1.5 T Siemens Sonata and GE Signa, as well as 3.0 T Siemens Trio. Closely matched acquisition sequences were used. However, repetition time and pulse sequence varied somewhat.
| Site | M021 (51) | M552 (92) | M554 (47) | M871 (44) |
|---|---|---|---|---|
| Scanner | Siemens Avanto | GE Signa | Siemens Trio | Siemens Sonata |
| Scanning sequence | GR | RM | IR\GR | GR |
| Sequence name | *fl3d1_ns | N/A | *tfl3d1_ns (19), tfl3d1_ns (28) | *fl3d1_ns (35), fl3d1_ns (9) |
| Slice thickness (mm) | 1.5 | 1.5 (20), 1.6 (38), 1.7 (31), 1.8 (3) | 1.5 | 1.5 |
| TR/TE (ms) | 12/4.76 | 20/6 | 2530/3.81 | 12/4.76 |
| Number of averages | 1 | 1 (2), 2 (90) | 1 | 1 |
| Magnetic field strength (T) | 1.494 | 1.5 | 2.8936 | 1.494 |
| Number of phase encoding steps | 256 | N/A | 256 | 288 |
| Percent phase field of view | 100 | 100 | 100 | 100 |
| Pixel bandwidth | 160 | 122 | 180 | 110 |
| Receiving coil | cp_head | 8ch_head | 8ch_head | 8ch_head |
| Acquisition matrix | 0 256 256 0 | 0 256 256 0 | 0 256 256 0 | 0 256 256 0 |
| Flip angle | 20 | 30 | 7 | 20 |
| Pixel spacing | 0.625 0.625 (11), 0.70313 0.70313 (40) | 0.625 0.625 (32), 0.66406 0.66406 (16), 0.70313 0.70313 (44) | 0.625 0.625 | 0.625 0.625 (42), 0.70313 0.70313 (2) |
- Each site is followed by the number of subjects that have been scanned using this setting, which also applies if a scanning setting varies within a site.
Preprocessing
The T1-weighted sMRI data were preprocessed at the Mind Research Network with Statistical Parametric Mapping 5 (SPM5, http://www.fil.ion.ucl.ac.uk/spm) using unified segmentation [Ashburner and Friston, 2005] in which image registration, bias correction, and tissue classification are performed using a single integrated algorithm. In this way, brains were segmented into gray matter, white matter, and cerebrospinal fluid and nonlinearly transformed into the ICBM152 standard space without Jacobian modulation. The resulting GMD images were re-sliced to 2 × 2 × 2 mm, resulting in 91 × 109 × 91 voxels. In the subsequent quality check, we excluded outliers whose correlations between the individual images and the across-subject average image were 4 standard deviations less than the mean. Based on this criterion, four subjects were excluded from the BIG data and no outliers were identified for the MCIC data. A mask was then generated (mean GMD > 0) to include only the segmented gray matter voxels, resulting in a total of 298,707 voxels for the BIG data and 292,998 voxels for the MCIC data. A voxelwise linear regression was performed to remove age and sex effects to avoid capturing associations majorly driven by these factors in the subsequent analysis. Furthermore, considering that the images were acquired with various scanning platforms, we employed the source-based-morphometry (SBM) approach [Xu et al., 2009] to investigate and eliminate the image variability introduced by scanning settings [Chen et al., 2014b]. Specifically, the data were decomposed into a linear combination of underlying sources using independent component analysis (ICA) [Amari, 1998; Bell and Sejnowski, 1995]. The loadings of each component were then assessed for associations with available scanning parameters. Components significantly affected by scanning settings were then identified and eliminated from the original data. Following this, nine scanning-related components were removed for the BIG data (Table 1) and eight scanning-related components removed for the MCIC data (Table 2). After correction, we did not observe any significant scanning effects. Finally, the corrected data were smoothed with an 8-mm full width at half-maximum Gaussian kernel and thresholded at mean GMD > 0.1 (resulting in 274,131 voxels) for the subsequent association analysis.
Genotyping
BIG
Saliva samples were collected from participants for DNA extraction. Genotyping was then conducted using the Affymetrix GeneChip SNP 6.0 array spanning more than 906,600 SNPs. The call rate threshold was set to 90%.
MCIC
DNA was extracted from blood samples. Genotyping for all participants was performed using the Illumina Infinium HumanOmni1-Quad assay spanning 1,140,419 SNP loci. BeadStudio was used to make the final genotype calls.
Imputation
To maximize the number of overlapping SNPs between two datasets, we imputed the MCIC data up to 5M SNPs using the MACH/Minimac pipeline [Howie et al., 2012] leveraging a large reference panel of the 1,000 Genomes data [Altshuler et al., 2012], as described in the ENIGMA protocol (http://enigma.ini.usc.edu/). We further excluded those imputed SNPs whose estimates of the squared correlations between imputed genotypes and true unobserved genotypes (rsq) were lower than 0.3, as recommended by the developer of the tool. To be consistent with the BIG data, the MCIC genotype data were obtained based on the continuous imputation data using the Genome-wide Complex Trait Analysis (GCTA) tool [Yang et al., 2011].
Data cleaning
PLINK software [Purcell et al., 2007] was used to perform a series of standard quality control procedures. SNPs and subjects were first examined for a genotyping rate threshold of 90%; SNPs were excluded if they showed deviation from Hardy–Weinberg Equilibrium with a threshold of 10−6 or if they failed to be missing at random with a threshold of 10−10; minor allele frequency cut-off was set to 0.01. Potential relatives were excluded if the Identity-By-Descent (IBD) was higher than 0.1875. For the BIG data, we first replaced the missing genotypes using haplotype genotypes of high linkage disequilibrium (LD) loci (correlation > 0.85). After the above procedures, missing genotypes were still observed in 134,607 out of 671,782 autosomal SNPs with missing ratios no greater than 0.05. We then replaced the remaining missing genotypes using the major alleles of individual loci. Discrete numbers were then assigned to the categorical genotypes: 0 for no minor allele, 1 for one minor allele, and 2 for two minor alleles. Finally, 666,019 common loci were genotyped or imputed in both the BIG and MCIC data. These loci were included for the association and validation analysis.
Association Analysis
 (1)
(1)
Flow chart of pICA-R. W1 and W2 denote the unmixing matrices of the two modalities, respectively. F1, F2, and F3 represent the objective functions based on which unmixing matrices are updated.
 (2)
(2) (3)
(3)The reference r is a binary vector with the same number of loci as the genomic data, where the selected referential loci are set to “1” and the rest are “0”s. This binary reference serves as a mask such that the distance between the component and reference vector is optimized particularly for the referential loci only. This design enables a semi-blind decomposition, where the presumed causal loci are constrained to be highlighted in the resulting component while the remaining loci are allowed to show their own importance driven by the data. For a full description of the mathematical details of pICA-R, we refer readers to the original publication [Chen et al., 2013].
Specifically in this work, the genetic references were derived from genes encoding heterotrimeric G-proteins. We leveraged the results from an imaging genetics study reporting associations between gray matter volume variations and SNPs in G-protein coding genes [Chavarria-Siles et al., 2013]. Among the seven genes identified in that previous work, GNA15, GNAO1, and GNB5 covered less than 10 SNPs in our data, therefore were not tested as references given that simulation results suggested an optimal reference of 20 true causal loci [Chen et al., 2013]. For the remaining four genes, GNG2, GNAQ, GNA14, and GNAL, we identified the corresponding SNPs in each gene and grouped neighboring SNPs with moderate correlations (r2 > 0.2, [Ripke et al., 2011]) into a cluster. The largest cluster within each gene was then used as the reference. This is because SNPs in one gene do not necessarily contribute simultaneously to only a single component, which is against the design of pICA-R. Instead, SNPs in one LD cluster are more likely to contribute to the same component and serve as good candidates for reference given that a marker allele in LD with the causal variant should show (by proxy) an association with the trait of interest [Stranger et al., 2011]. In this way, we tested four references hosted by GNG2, GNAQ, GNA14, and GNAL.
Like infomax ICA, pICA-R requires estimating the number of components before data decomposition. Minimum description length (MDL) [Rissanen, 1978] is commonly used for the imaging modality to select the component number yielding the most efficient representation of the original data. However in the SNP data, most genetic factors account for small amounts of variance, except for those related to population structure. Thus, MDL is much less applicable to select the component number such that the major variance in the data is retained. Instead, we chose to estimate the component number based on components’ consistency, to obtain the most stable data decomposition [Chen et al., 2012].
Validation
 (4)
(4)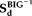 represents the pseudo inverse of the component matrix extracted from the BIG data.
represents the pseudo inverse of the component matrix extracted from the BIG data.
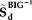 and
and
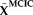 denote respectively the submatrices of
denote respectively the submatrices of
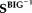 and the MCIC observed data, corresponding to the top voxels or SNPs presenting relatively stronger correlations with the loadings. AMCICrepresents the loaing matrix estimated through projecting the top voxels or SNPs. We expect that the loadings obtained in this way should more accurately reflect the effects of the most important markers. Finally, the correlations between the projected loadings were calculated to assess whether the sMRI-SNP association identified in the BIG data remained significant in the MCIC data.
and the MCIC observed data, corresponding to the top voxels or SNPs presenting relatively stronger correlations with the loadings. AMCICrepresents the loaing matrix estimated through projecting the top voxels or SNPs. We expect that the loadings obtained in this way should more accurately reflect the effects of the most important markers. Finally, the correlations between the projected loadings were calculated to assess whether the sMRI-SNP association identified in the BIG data remained significant in the MCIC data.Analyzing Components
To understand the influences on cognition, the identified SNP and sMRI components were further investigated for associations with available phenotypic data, which was the reversal learning score collected for 599 out of 1,256 BIG subjects. In addition, top contributing voxels of the identified sMRI component were mapped to the Talairach atlas [Lancaster et al., 1997, 2000] for the involved brain regions. Meanwhile, top contributing SNPs were annotated and the hosting genes were sent for Ingenuity Pathway analysis (IPA, http://www.ingenuity.com/) to reveal the genetic architecture.
RESULTS
The number of components was estimated to be 10 and 11 for the sMRI and SNP data, respectively. Among all the tested references, the one derived from GNA14 elicited a significant sMRI-SNP association (r = −0.16, P = 2.27 × 10−8). This exceeded the conservative Bonferroni threshold of 1.14 × 10−4 which corrected for all the tested four genetic references and the combinations of all extracted components (10 sMRI × 11 SNP) in each run. The reference comprised 22 out of 83 SNPs from GNA14, as listed in Supporting Information Table S1. These SNPs are in moderate LD, presenting a mean r-square of 0.54, and distanced by an average of 2,557 base pairs. The sMRI-SNP association remained significant, exhibiting a correlation of −0.16 (P = 2.34 × 10−8) when the SNP component was controlled for age and sex, as shown in Figure 2a. Besides, the SNP-sMRI association remained significant (P = 3.30 × 10−7) when the top three principal components of the SNP data were further included as covariates [Ripke et al., 2011], indicating that the observed association was not likely attributable to ancestral background. The associated sMRI component was thresholded at |Z| > 2 for top voxels. The number of top SNPs was determined based on the absolute values of the component z-scores using a more conservative linear fitting approach (for details see Supporting Information Figure S1), yielding a total of 2,000 top SNPs corresponding to |Z| > 3.37. We then projected the top voxels and SNPs to the MCIC data, and the resulting loadings again yielded a significant correlation of −0.25 (P = 0.02) when controlling for age and sex, as shown in Figure 2b. To be noted, the projected association was robust to the threshold selection, remaining significant with a number of top SNPs ranging from 1,000 to 2,500. No association was observed between the identified sMRI/SNP component and the reversal learning score.

Scatter plots of (a) the single nucleotide polymorphism (SNP) and sMRI loadings identified in the BIG data; and (b) the SNP and sMRI loadings obtained from the MCIC data through projection.
The top contributing voxels of the sMRI component were further mapped to nearest gray matter to obtain the Talairach atlas labels [Lancaster et al., 1997, 2000]. Figure 3 and Table 3 show the spatial map and the mapped regions, respectively. It can be seen that the identified brain network comprised superior and medial frontal gyri, precuneus, and cingulate gyrus. For the SNP component, Supporting Information Table S2 provides a summary of the top contributing SNPs, including chromosome, base pair position, corresponding gene, minor allele, and component z-score. From the GNA14 reference, five SNPs were identified as top contributors to the associated SNP component, including rs10869927_G (“G” for the minor allele), rs10781441_T, rs12684903_A, rs2889774_T, and rs7047853_G. Figure 4 shows a Manhattan plot of weights of loci for the identified SNP component, where clusters are marked if they span more than five SNPs with maximum normalized weights greater than 5. Out of the top 2,000 SNPs, 803 are intragenic and mapped to 272 unique genes, including another G-protein, GNA12, and two G-protein-coupled receptors, GPR113 and GPR133. IPA revealed that these 272 genes were significantly enriched in a number of canonical pathways, including synaptic long-term potentiation (LTP) and depression (LTD), axonal guidance, protein kinase A (PKA), CREB, and nNOS signaling, as listed in Table 4. In particular, all the pathways highlighted in bold remained significantly enriched when the number of top SNPs was adjusted from 1,000 to 2,500, indicating a relatively consistent genetic architecture. We also explored through IPA potential interactions between selected genes of interest, including GNA12, GNA14, GRM1, PRKCD, CAMK2B, and PDIA3. The constructed network is illustrated in Supporting Information Figure S2.

Spatial map of the identified sMRI component (|Z| > 2). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]

Manhattan plot for the identified single nucleotide polymorphism (SNP) component. The horizontal line indicates the threshold at |Z| > 3.37 for selection of top contributing SNPs. Clusters are marked if they span more than 5 SNPs with maximum normalized weights greater than 5. The GNA14 cluster is also marked. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
| Brain region | Brodmann area | L/R volume (cm3) | L/R random effects, max Z (x, y, z) |
|---|---|---|---|
| Medial frontal gyrus | 6, 8, 32, 9, 10 | 13.9/8.7 | 6.53 (0,16,47)/5.79 (2,−6,65) |
| Superior frontal gyrus | 6, 8, 9, 10 | 12.3/9.6 | 7.15 (0,14,53)/6.01 (2,14,53) |
| Precuneus | 7, 19 | 8.0/6.0 | 6.44 (0,−47,63)/6.29 (2,−47,63) |
| Paracentral lobule | 5, 4, 6, 31, 7 | 5.2/4.1 | 7.01 (0,−41,65)/6.63 (2,−43,63) |
| Cingulate gyrus | 24, 32, 31 | 4.0/2.0 | 5.59 (0,2,48)/4.79 (2,18,43) |
| Postcentral gyrus | 5, 7, 4, 3, 2 | 3.5/3.5 | 6.75 (−2,−41,65)/6.81 (2,−41,65) |
| Middle frontal gyrus | 6, 8 | 2.7/2.9 | 2.88 (−28,1,61)/3.38 (28,5,59) |
| Ingenuity canonical pathways | Molecules | P-value |
|---|---|---|
| Neuropathic pain signaling in dorsal horn neurons | NTRK2, KCNQ2, GRM1, PDIA3, KCNQ3, PRKCH, CAMK2B | 6.76 E−04 |
| Inositol pyrophosphates biosynthesis | IP6K3, PPIP5K1 | 4.37 E−03 |
| Histidine degradation III | UROC1, AMDHD1 | 5.75 E−03 |
| Histidine degradation VI | UROC1, AMDHD1 | 7.41 E−03 |
| Synaptic long-term potentiation | GRM1, PDIA3, PRKCH, GNA14, ADCY8, CAMK2B | 7.59 E−03 |
| Protein kinase A signaling | EPM2A, PTPRK, PTPRD, PTPRJ, PDIA3, PDE8B, PRKCH, ADCY8, EYA2, PTPRT, NTN1, CAMK2B | 8.51 E−03 |
| Axonal guidance signaling | PDIA3, GNA12, GNA14, ADAMTS2, NTN1, SRGAP3, NTRK2, NTRK3, EFNA5, PAK7, PRKCH, WNT5A, GLIS1 | 1.05 E−02 |
| CREB signaling in neurons | GRM1, PDIA3, GNA12, PRKCH, GNA14, ADCY8, CAMK2B | 1.32 E−02 |
| Synaptic long-term depression | GRM1, PDIA3, GNA12, PLA2R1, PRKCH, GNA14 | 1.62 E−02 |
| Sulfate activation for sulfonation | PAPSS2 | 2.95 E−02 |
| Formaldehyde oxidation II (glutathione-dependent) | ESD | 2.95 E−02 |
| Glutamine degradation I | GLS | 2.95 E−02 |
| nNOS signaling in neurons | DLG2, PRKCH, NOS1AP | 3.09 E−02 |
| Endothelin-1 signaling | PDIA3, GNA12, PLA2R1, PRKCH, GNA14, ADCY8 | 3.89 E−02 |
| GNRH signaling | PAK7, PRKCH, GNA14, ADCY8, CAMK2B | 4.07 E−02 |
| RhoGDI signaling | GNA12, ARHGAP12, CDH18, PAK7, GNA14, CDH13 | 4.37 E−02 |
| Methionine salvage II (Mammalian) | BHMT2 | 4.37 E−02 |
DISCUSSION
In this work, we applied pICA-R to explore genomic basis of brain structure in a homogeneous cohort of healthy Caucasians. G-proteins arose as potential references given that they are implicated in neural development. A previous univariate study [Chavarria-Siles et al., 2013] provided more direct evidence for associations between G-protein SNPs and regional gray matter variations. We then leveraged these results and derived four genetic references which were then investigated in the semi-blind multivariate framework. In the BIG dataset which included 1,256 subjects, a GNA14 reference elicited a significant SNP-sMRI association (r = −0.16, P = 2.34 × 10−8), while the other three tested references did not yield any significant finding. As discussed by Chen et al. [2013], references derived from univariate models do not always yield significant findings in multivariate analyses. This might be due to various factors. One possibility is that the reference SNPs may have heterogeneous effects and contribute to different components. Or the related network as a whole may not be well represented in this specific dataset.
Using a semi-blind multivariate approach, we identified extended SNP and sMRI components compared to the previous work by Chavarria-Siles et al. The identified brain network consisted of medial frontal gyrus, superior frontal gyrus, precuneus, and cingulate regions. To be noted, the medial frontal cortex was also identified by the Chavarria-Siles et al. that employed 532 BIG subjects and used a different method. Regarding the genetic modality, five reference SNPs from GNA14, as expected, were identified as top contributors to the associated SNP component. The SNP highlighted in Chavarria-Siles et al. (rs4745639) was not included in the reference, however it showed moderate LD with rs2889774 and rs10869927 (r2 of 0.15 and 0.14, respectively), and all contributed with negative component weights. The elicited SNP component was enriched in pathways related to neuronal functions, as listed in Table 4. More importantly, the identified association was further replicated in the independent MCIC dataset. Compared to univariate analyses, pICA-R assesses multiple variables for the aggregate effect. For the SNP modality, pICA-R is well posed to model polygenicity and to capture the additive effect of multiple SNPs with moderate individual effects [Chen et al., 2013; Polderman et al., 2015]. For the sMRI modality, pICA-R extracts patterns of variations shared by regional and distant voxels [Xu et al., 2009]. And imaging genetic associations are then evaluated between the SNP and sMRI components’ loadings. Thus, our results delineate a genomic basis underlying a proportion of the GMD variation in the highlighted frontal and parietal regions.
The spatial map of the identified sMRI component clearly highlighted the midline of brain, which is frequently implicated in neural development, ageing, and cognition [Frangou et al., 2004; Lyuksyutova et al., 2003; Shaw et al., 2006; Sowell et al., 2003]. Prefrontal cortex is known to be involved in complex cognitive behavior and decision making [Koechlin and Hyafil, 2007; Koechlin et al., 2003]. Precuneus plays a key role in highly integrated tasks, including episodic memory retrieval, self-referential processes, and consciousness [Laureys et al., 2004; Lundstrom et al., 2005; Ochsner et al., 2004]. Cingulate cortex is an integral part of the limbic system and active in a variety of cognitive functions such as emotion, learning and memory [Bush et al., 2000]. More interestingly, these regions are also among those showing relatively significant structural variations for different age and intelligence groups. A VBM study demonstrated positive correlations between IQ and GMD in voxel clusters distributed along the brain midline, including frontal cortex, cingulate, and precuneus [Frangou et al., 2004]. At superior and medial frontal gyri, the trajectories of cortical thickness change with age are found to be different between the superior and the high/average intelligence groups [Shaw et al., 2006]. Besides, axons extend on either side of the midline (anterior–posterior axis) to form longitudinal tracts and the midline cells are crucial for guiding axon outgrowth [Lyuksyutova et al., 2003]. These observations suggest that the identified brain network captures regional GMD co-variations that can play a role in cognitive abilities and might reflect inter-individual differences in brain development.
The SNP component was overrepresented in a number of pathways communicating with each other and with G-proteins. From those shown in Table 4, LTP and LTD are two forms of synaptic plasticity that affect signal transmission between neurons and have been widely studied to understand the mechanisms underlying learning and memory [Martin et al., 2000; Neves et al., 2008]. It is believed that glutamate receptors are major triggers for the induction of LTP and LTD, where PKA, protein kinase C (PKC), calcium/calmodulin-dependent protein kinase II (CaMKII) can all play a role [Collingridge et al., 2004]. CREB is majorly activated by cAMP through PKA [Delghandi et al., 2005]. In particular, a behavioral experience can elicit synaptic activities which activate CREB, inducing expression of molecules contributing to consolidating changes in synaptic strength [Benito and Barco, 2010]. Axon guidance strongly relies on a number of cue molecules, among which netrins are the most well understood. It has been demonstrated that netrins retain the function of attracting axons toward the brain midline while also repelling some axons, and responses to the guidance cue netrin-1 (NTN1) are sensitive to levels of cAMP or PKA activity [Dickson, 2002]. Neuronal nitric oxide synthase (nNOS) is a biosynthetic enzyme functioning in several types of synaptic plasticity, including LTP and LTD [Bon and Garthwaite, 2003; Nelson et al., 1995]. The phosphorylation of nNOS is regulated by kinases and phosphatases such as PKA, PKC, and CaMKII [Zhou and Zhu, 2009]. It is noteworthy that G-proteins are involved in the signaling cascades of all these cellular machineries, including glutamate, cAMP, PKA, PKC, CREB, and CaMKII [Neves et al., 2002].
Among the enriched pathways, overlaps of genes were observed, including GRM1, PRKCH, GNA12, GNA14, and CAMK2B. GRM1 encodes a metabotropic, G protein-coupled receptor for glutamate, the major excitatory neurotransmitters in the central nervous system [Hermans and Challiss, 2001]. A total of 10 SNPs from GRM1 were identified as top contributors to the component in our analysis. Nine out of these 10 SNPs contributed positively (see Supporting Information Table S2 for details). Note that the SNP-sMRI correlation was negative, indicating that subjects with higher loads of the SNP component carried relatively lower loads of the sMRI component. Also, the highlighted brain regions presented positive component weights, indicating that subjects with higher loads of the sMRI component had higher regional GMD. Thus, for these SNPs presenting positive component weights, our finding suggested that, overall, subjects carrying more minor alleles at these loci showed reduced GMD in the highlighted brain regions. Only one out of the 10 top SNPs in GRM1 (rs854144_T) contributed with a negative weight, indicating more minor alleles found in subjects exhibiting increased regional GMD. PRKCH is another molecule of great importance as it encodes a PKC subtype. PKCs phosphorylate a wide range of protein targets involved in brain functioning. For instance, phosphorylation of GluR1 by PKC is demonstrated to be critical for LTP expression [Boehm et al., 2006]. In our analysis, one single SNP rs1957902_G was identified from PRKCH, presenting a negative weight. GNA12 and GNA14 fall into the G-protein subfamilies of G12 and Gq. GNA12 has been indicated to negatively regulate cell adhesion through interacting with cadherin [Meigs et al., 2002]. GNA14 is among the genes which were found to exhibit developmental expression variations in the cortex of a marmoset animal model, and linked to axonal guidance [Sasaki et al., in press]. In the present study, rs2258960_T and rs2644311_T were identified from GNA12 and presented positive weights. For GNA14, rs10869927_G and rs2889774_T showed negative weights while rs10781441_T, rs12684903_A, and rs7047853_G showed positive weights. CAMK2B encodes a Ca2+/calmodulin-dependent protein kinase involved in calcium signaling. The expression of this gene has been found to be disrupted in Alzheimer's disease [Antonell et al., 2013; Liang et al., 2008]. Three SNPs, rs12702075_A, rs10281178_C, and rs3934888_G, were identified from CAMK2B, all exhibiting negative weights.
Both the imaging and genetic findings from our analysis link strongly to neural development, indicating that the observed GMD variations might be traced back to developmental processes under control of genetic factors. It is well acknowledged that genetic components underlie anatomic variations in healthy and diseased human brains [Andersen, 2003; Giedd, 2005; Sotelo, 2004]. Direct associations between genetic variants and quantitative structural measures have also been demonstrated in large-scale studies [Bis et al., 2012; Stein et al., 2012]. Compared with previous works using univariate approaches, our results delineate relationships between particular genetic pathways and regional structural variations, emphasizing that these G-protein-related PKA, CREB, and nNOS mechanisms play a critical role in brain development which might ultimately lead to GMD differences. On the other hand, it should be noted that brain structure is the final manifestation of a complex interplay between multiple genetic and environmental factors. Our analysis captured a part of the story.
The current study benefited from a large homogeneous discovery sample which enhances statistical power. In addition, the employed multivariate approach assessed a group of variables whose aggregate effects would be more prominent compared to those of individual variables. Leveraging prior knowledge further improved the chance of pinpointing factors of interest in a large complex dataset. Most importantly, the imaging genetic association identified from the discovery sample was replicated in an independent cohort, indicating a low possibility of false positive results. On the other hand, one major limitation of this study lies in the different scanning platforms employed for imaging data collection, which can be a potential confound. To address this issue, we carefully explored all the scanning settings and eliminated from the imaging data the variance most likely induced by scanning discrepancies based on a linear model. Nonlinear scanning effects might still exist for which we do not have any knowledge at present. However, such effects were not expected to significantly contribute to the identified imaging genetic association given the linear decomposition of ICA. Another limitation is that it is not clear yet how the identified components are related to behavior based on the available data. The identified local GMD variations have been implicated in intelligence studies; however we did not collect IQ data in this project. Instead we investigated whether this imaging trait was correlated with the available reversal learning scores and found no significant associations. Further work needs to be done to better understand how the genetic and GMD variations might lead to differences in behavior. Besides, it should be noted that SNPs in high LD would exhibit comparable effects in our analysis. Therefore, SNPs might be identified due to tagging true causal variants. However, the identified genes and pathways should be unchanged. Finally, following the design of pICA-R, we only tested the largest cluster as a genetic reference for each candidate gene in this work, ignoring other smaller clusters which might also be biologically informative. This could be tackled with an extended approach, parallel ICA with multiple references [Chen et al., 2014a], which explores potential convergence of functional influences among genes. We plan to conduct such an analysis in a future study.
In summary, we performed a guided exploration in the present study, where a reference derived from the GNA14 gene managed to elicit a genetic component significantly associated with an sMRI component in a semi-blind multivariate analysis. The associated genetic component highlighted several neural signaling pathways which appear to interact with each other and are active in synaptic plasticity and axonal guidance. Meanwhile, the identified brain network comprised regions recruited in various cognitive processes and robustly implicated in brain maturation and intelligence. Collectively, our study suggests a key role of G-proteins in the genetic architecture underlying normal GMD variations in frontal and parietal regions. We speculate that the observed GMD variations partially result from differential genetic modulation of brain development, though future longitudinal studies are needed to dissect genetic contribution to trajectories of anatomic changes in developing brains.
ACKNOWLEDGMENTS
This work makes use of the BIG (Brain Imaging Genetics) database, first established in Nijmegen, The Netherlands, in 2007. This resource is now part of Cognomics (www.cognomics.nl), a joint initiative by researchers of the Donders Centre for Cognitive Neuroimaging, the Human Genetics and Cognitive Neuroscience departments of the Radboud University Nijmegen Medical Centre and the Max Planck Institute for Psycholinguistics in Nijmegen. The Cognomics Initiative is supported by the participating departments and centres and by external grants, i.e. the Biobanking and Biomolecular Resources Research Infrastructure (Netherlands) (BBMRI-NL), the Hersenstichting Nederland, and the Netherlands Organisation for Scientific Research (NWO). The authors thank all persons who kindly participated in the BIG research. The Board of the Cognomics Initiative consists of Barbara Franke, Simon Fisher, Guillén Fernandez, Peter Hagoort, Han G. Brunner, Jan Buitelaar, Hans van Bokhoven and David Norris. The authors would also like to thank the University of Iowa Hospital, Massachusetts General Hospital, the University of Minnesota, the University of New Mexico, and the Mind Research Network staff for their efforts in data collection, preprocessing, and analyses.

