

Flexible and Robust Methods for Rare-Variant Testing of Quantitative Traits in Trios and Nuclear Families
Contract grant sponsor: National Institutes of Health; Contract grant number: HG007508.
ABSTRACT
Most rare-variant association tests for complex traits are applicable only to population-based or case-control resequencing studies. There are fewer rare-variant association tests for family-based resequencing studies, which is unfortunate because pedigrees possess many attractive characteristics for such analyses. Family-based studies can be more powerful than their population-based counterparts due to increased genetic load and further enable the implementation of rare-variant association tests that, by design, are robust to confounding due to population stratification. With this in mind, we propose a rare-variant association test for quantitative traits in families; this test integrates the QTDT approach of Abecasis et al. [Abecasis et al., 2000a] into the kernel-based SNP association test KMFAM of Schifano et al. [Schifano et al., 2012]. The resulting within-family test enjoys the many benefits of the kernel framework for rare-variant association testing, including rapid evaluation of P-values and preservation of power when a region harbors rare causal variation that acts in different directions on phenotype. Additionally, by design, this within-family test is robust to confounding due to population stratification. Although within-family association tests are generally less powerful than their counterparts that use all genetic information, we show that we can recover much of this power (although still ensuring robustness to population stratification) using a straightforward screening procedure. Our method accommodates covariates and allows for missing parental genotype data, and we have written software implementing the approach in R for public use.
Introduction
The emergence of next-generation sequencing technology, along with the development of the exome chip, has led many investigators to study the role of rare genetic variation in complex human traits. Rather than analyze rare variants individually, many statistical approaches for rare-variant association mapping employ grouping strategies that aggregate rare variants in a gene or region for analysis to improve power. These approaches can be broadly categorized as either burden tests that collapse grouped rare variants into a single aggregate variable that is then regressed on phenotype [Li and Leal, 2008; Madsen and Browning, 2009; Morris and Zeggini, 2010; Zawistowski et al., 2010], kernel tests that relate phenotype to rare variants in a region as a function of a variance component (SKAT), [Wu et al., 2011], and unified tests that combine burden and kernel tests together (SKAT-O), [Lee et al., 2012]. Burden tests are preferred when a region harbors rare causal variants that all act in the same direction on phenotype (all protective or all deleterious) whereas kernel tests are optimal when a region harbors rare causal variants that act in different directions on phenotype [Wu et al., 2011].
Although these rare-variant methods generally have improved power compared to tests of individual rare variants, almost all of these tests are restricted to case-control or population-based study designs and cannot be used in family-based studies. Family-based designs have several advantages over population-based designs in that they enable the use of statistics that, by design, are robust to confounding due to population stratification. Family designs also can solve genetic problems that are hard to answer in population-based studies. For example, sequencing the parents of affected subjects can identify de novo mutations and also allow the study of rare homozygous genotypes, which are difficult to find in population-based designs [Do et al., 2012]. Families are also attractive to study because they often provide increased genetic load for a disease or trait: although carriers of a minor risk allele will be hard to sample in the general population, they are more likely to be found in families of probands [Zollner, 2012]. Finally, family studies allow the study of the segregation pattern of complex disease [Ott et al., 2011]. Because of these appealing features and the fact that there are many familial samples from past linkage studies, family-based resequencing studies are gaining in popularity. Several recent studies have identified disease-associated rare variants through family-based designs, including rare-variants associated with multiple sclerosis [Ramagopalan et al., 2011], simplex autism [Krumm et al., 2013], dilated cardiomyopathy [Norton et al., 2011], and Alzheimer's disease [Cruchaga et al., 2012].
Recently, a few methods have been proposed for rare-variant association testing in families. Schaid et al. [2013] developed a method for complex traits that accounts for relatedness among study subjects. Their method took a retrospective view of the sample, which assumes that the outcome is fixed while the genotype is random, and is particularly appealing for the analysis of datasets that are collected under nonrandom ascertainment (such as those collected for linkage studies). Chen et al. [2013] developed a rare-variant test for quantitative traits in families by extending kernel-machine methods [Kwee et al., 2008; Wu et al., 2011] to pedigree analysis by inserting a random familial effect due to shared polygenes within the modeling framework; a similar idea was employed by Schifano et al. [2012] and Oualkacha et al. [2013]. Jiang and McPeek [2014] adopted a similar strategy to extend the SKAT-O [Lee et al., 2012] method to family studies of quantitative traits. Although these methods adjust for kinship in family studies, they do not consider potential bias caused by population stratification. Population stratification can lead to substantially inflated false-positive rates in sequencing studies of rare variants [Epstein et al., 2012; Jiang et al., 2013; Liu et al., 2013], and standard GWAS approaches to correct for such stratification (such as principal components or EMMAX) [Kang et al., 2010] may not be effective when applied to rare variants [Mathieson and McVean, 2012]. Therefore, a rare-variant association test that maintains validity in the presence of such stratification is needed. Ionita-Laza et al. [2013] proposed such a method based on the family-based association test (FBAT) framework. Although this method is robust to population stratification, it ignores between-family information that could perhaps be exploited to boost power. Fang et al. [2012, 2013] used between-family information for this purpose in an adaptive rare-variant association test for quantitative traits; however, the procedure requires computationally intensive permutations for inference, so it is unclear whether the approach is scalable to large-scale resequencing efforts.
In this paper, we propose a novel two-stage method for rare-variant analysis of quantitative traits in trios and nuclear families. The approach is based on the QTDT (quantitative transmission disequilibrium test) framework of Abecasis et al. [2000a] for SNP association mapping. The QTDT framework decomposes the observed individual genotypes into between-family and within-family components. The within-family component is robust to population stratification, although the between-family component is sensitive to the phenomenon. In this paper, we calculate the within-family component for each rare variant in a region, and then integrate these components within the kernel procedure KMFAM of Schifano et al. [2012], which was previously developed for SNP-set association testing of quantitative traits in families. Specifically, within KMFAM, we create a kernel matrix based on the within-family component, and then use this kernel matrix to test for association with phenotype using a modified score statistic. By using the within-family component only, our rare-variant association test for quantitative traits is robust to confounding due to population stratification. Also, the approach calculates P-values analytically rather than via resampling and is thus scalable to exome sequencing and whole-genome resequencing studies. Because the approach relies on a kernel framework, it also preserves power when a region contains a mixture of trait-increasing and trait-decreasing variation. The approach also allows for covariates and, for nuclear families, can be implemented when phenotype and genotype data on parents are missing, so it can be applied in the study of quantitative traits related to late-onset diseases.
A potential drawback of using only within-family information for analysis is that power is reduced by ignoring the (sensitive) between-family information within the analysis [Ionita-Laza et al., 2013]. However, borrowing ideas from Purcell et al. [2005] and Van Steen et al. [2005], we propose using between-family information as a screening tool to identify the most interesting regions (based on the magnitude of the P-value for the region) that merit further investigation. We then apply our within-family test to only these top regions, thereby reducing the multiple-testing burden (compared to within-family testing of all regions) and potentially gaining power. We note that the first stage of the analysis (using the between-family information) is independent of the second stage (which uses orthogonal within-family information). We also note that, by using within-family information in the second stage, our approach is still robust to confounding due to population stratification.
In subsequent sections, we first describe the KMFAM procedure and then, for rare variants, discuss how we integrate the QTDT framework into the model to make the method robust to population stratification. We next describe our screening procedure to improve power. We then evaluate our approaches using simulated sequence data in trios and nuclear families and show how screening can improve power of within-family testing although maintaining an appropriate type I error rate, even under population stratification. Finally, we summarize our method and discuss potential extensions.
Materials and Methods
Notation and KMFAM Model
 denote the quantitative outcome for the jth individual in the ith family, where i = 1, 2, 3…N and j = 1, 2, … ni. We define
denote the quantitative outcome for the jth individual in the ith family, where i = 1, 2, 3…N and j = 1, 2, … ni. We define  as a c ×1 vector that represents the covariates for the jth individual in the ith family and further define Gij as an s ×1 vector that represents the genotypes of the s rare variants for each subject (where each rare-variant genotype is coded as the number of copies of the rare allele the subject possesses at each site). We assume that the outcome, Yij, follows a multivariate normal distribution with mean and variance defined through the model:
as a c ×1 vector that represents the covariates for the jth individual in the ith family and further define Gij as an s ×1 vector that represents the genotypes of the s rare variants for each subject (where each rare-variant genotype is coded as the number of copies of the rare allele the subject possesses at each site). We assume that the outcome, Yij, follows a multivariate normal distribution with mean and variance defined through the model:
 (1)
(1) , which will have low power.
, which will have low power. , where Φi is the kinship matrix for family i and
, where Φi is the kinship matrix for family i and 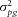 is the variance due to the effect of polygenes. We also define
is the variance due to the effect of polygenes. We also define  as the random error term. From model (1), we calculate the variance of outcome as
as the random error term. From model (1), we calculate the variance of outcome as
 (2)
(2) (3)
(3) and
and 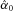 as the estimates of V in (2) and α in (1) under the null. Further, we define a projection matrix
as the estimates of V in (2) and α in (1) under the null. Further, we define a projection matrix  , such that
, such that  . Thus, under the null, we have
. Thus, under the null, we have
 (4)
(4) are eigenvalues of
are eigenvalues of 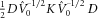 , here
, here  . As
. As  are independently and identically distributed random variables, Q is distributed as an asymptotic mixture of chi-square distributions, and the P-values can be calculated using the Davies method [Davies, 1980].
are independently and identically distributed random variables, Q is distributed as an asymptotic mixture of chi-square distributions, and the P-values can be calculated using the Davies method [Davies, 1980].Robust Rare-Variant Association Test

Once we obtain the between-family component, we then construct the within-family component, Wij, by subtracting the between-family component from the observed genotype such that Wij = Gij − Bij.
By design, association analyses of complex traits that base inference on the within-family component Wij, are robust to population stratification. Based on this observation, we can construct a robust rare-variant association test for trios and nuclear families by replacing the observed genotypes G in the kernel matrix K described in (2) with their corresponding within-family components W. We then construct the score statistic Q in (3) as before to derive our robust association test.
Screening Procedure
Although the QTDT framework ensures the robustness of our proposed score test to potential confounding due to population stratification, the discarding of between-family information when confounding due to population stratification is not an issue can lead to sizable power loss compared to use of the observed genotype. In attempts to restore the power of our within-family association test to levels anticipated when using observed-genotype information, we suggest a two-stage screening approach that uses both the within- and between-family rare-variant information. In the first stage, we use between-family information to screen and identify the top regions for follow up. If parental phenotype and genotype information are available, we carry out the first stage by performing the SKAT [Wu et al., 2011] test on parents only, and then select a subset of regions for follow-up investigation based on smallest P-values. If parental information is unavailable, we instead conduct the first-stage screening by applying KMFAM to the outcomes and between-family components of the offspring. In the second stage, we construct the robust test (using the within-family components calculated for the offspring) only on those top regions selected from the first stage. By only testing a reduced number of regions in the second stage using the within-family component, we reduce the number of robust tests that are conducted thereby reducing the multiple-testing burden and increasing power. As discussed in Abecasis et al. [2000a], the between-family and within-family components are orthogonal to each other, such that the first-stage and second-stage tests are independent.
Type I Error Simulations
We evaluated the type I error and power of our approach using simulated sequencing data. We used cosi [Schaffner et al., 2005] to simulate sequence data for a pool of 5000 European and 5000 African haplotypes, each of length 30 kb. Rare variants were defined as variants with a minor-allele frequency greater than 0% and less than 3% in the region. To simulate family data, we randomly paired subjects within each population and simulated offspring by sampling one haplotype from each parent. When considering nuclear families with 2 or more offspring, we performed simulations for the situation where all parental information is available, as well as where 20–100% parental information is missing.
 (5)
(5) is an indicator variable that is 1 if the subject is African and 0 otherwise, and all other terms are the same as defined in model (1). We specified fij and εij such that the overall trait heritability was 0.35. To induce confounding due to population stratification in our simulations, we first assumed our sample consisted of a mixture of European and African families, with the percentage of European families ranging from 25% to 75%. We then assumed a value of γ in model (5) that ranged from 0 (no confounding due to population stratification) to 3 (extreme confounding due to population stratification).
is an indicator variable that is 1 if the subject is African and 0 otherwise, and all other terms are the same as defined in model (1). We specified fij and εij such that the overall trait heritability was 0.35. To induce confounding due to population stratification in our simulations, we first assumed our sample consisted of a mixture of European and African families, with the percentage of European families ranging from 25% to 75%. We then assumed a value of γ in model (5) that ranged from 0 (no confounding due to population stratification) to 3 (extreme confounding due to population stratification).Power Simulations
To estimate power, we simulated a region of 300 kb, divided into 10 nonoverlapping regions of 30 kb each, and selected one region at random as causal (the other nine regions are assumed to be independent of outcome). To generate trait data for each subject based on the causal region, we used the idea of [Wu et al. 2011] and assumed a certain percentage (5% or 15%) of rare variants (defined as variants with a MAF less than 3%) in the region influenced the outcome, with the effect size of a causal variant defined as 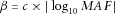 , where we varied the constant c among values between 0.4 and 0.6. We then included these effects due to rare variants within model (5) to simulate the outcome. To keep power at a reasonable range for the 300 kb region, we fixed γ at 0.25 for power simulations under stratification. As with the null simulations, we assumed the trait heritability was 0.35.
, where we varied the constant c among values between 0.4 and 0.6. We then included these effects due to rare variants within model (5) to simulate the outcome. To keep power at a reasonable range for the 300 kb region, we fixed γ at 0.25 for power simulations under stratification. As with the null simulations, we assumed the trait heritability was 0.35.
Results
Type I Error
We first performed type I error rate simulations on parent-offspring trios to demonstrate that population stratification can lead to spurious association with quantitative traits in families. Figure 1 presents type I error results for two methods: our robust rare-variant approach that uses within-family information from the offspring only and a SKAT test of rare-variant association that uses the observed offspring genotype (constituting both the within- and between-family components). For these simulations, simulated datasets consisted of 500 trios where 50% are of European descent and the remaining 50% are of African descent. When the mean trait difference between European and African populations is 0 (such that there is no confounding due to population stratification), both the within-family test and observed-genotype test had appropriate type I error. However, when we induced confounding due to population stratification by assuming a nonzero mean trait difference between Africans and Europeans, we found the standard SKAT test using the observed genotype had inflated type I error. Our robust rare-variant association test, in contrast, maintained the proper type I error rate under confounding.

We next performed another set of type I error simulations, where we assumed datasets consisting of 500 nuclear families each with two children. We varied the proportion of nuclear families that were of European origin between 25% and 75% and assumed the mean trait difference between African and European samples to be 2 (thereby inducing confounding due to population stratification). We further assumed the proportion of nuclear families within each dataset that was missing parental genotype information ranged from 0% to 100%. In our first set of simulations (shown in Figure 2, we studied the type I error rates of methods assuming examination of the 30 kb region in its entirety. We compared the type I error rates using the observed genotype information in the offspring only, accounting for kinship (which corresponds to the KMFAM test of Schifano et al. [2012] as well as the test of Chen et al. [2013]), as well as using our robust rare-variant association test that relies only on the within-family information in the offspring. Our results indicated that rare-variant association tests using observed genotype information led to considerable inflation in type I error rates across different simulation models, whereas our robust within-family association test remained valid in all situations. The validity of the robust rare-variant association test was confirmed both when parental genotype information was available on all participants, as well as when such genotype information was completely absent in the dataset. Thus, for late-onset diseases in which parental information might not be available, our method is still robust to population stratification.

We performed a final set of type I error simulations for nuclear families of size two under our proposed screening scheme where, in this instance, we split the 300 kb region into 10 nonoverlapping regions, each of size 30 kb. Using between-family information, we identified a subset of regions for follow up (based on P-value) that we then investigated further using the within-family component. Our results are shown in Figure 3. Overall, our results show that our screening procedure (conducted using either parental information or between-family information in siblings, if parents are not available) preserved type I error across models, with differing missing parental information as well as different proportions of regions that were then followed up using within-family information. These results demonstrate that our screening procedure maintains appropriate type I error, even when there is confounding due to population stratification, due to the fact that the between-family component and within-family component of the offspring genotype are orthogonal to one another.

Power
In the previous section, we showed that our robust rare-variant association test that uses the within-family component remains valid in the presence of population stratification. We next studied the power of our proposed robust test to detect association with a trait under various trait-influencing models. We assumed either 5% or 15% of rare variants in a region were causal and assumed the effect size of such causal variants was 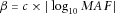 , where c ranged from 0.4 to 0.6. We first compared the power of our robust within-family association test to the standard observed-genotype test considered by Chen et al. and Schifano et al. under models with no population stratification (to ensure the power of the observed-genotype test was valid). We generated sequence and trait data on 500 nuclear families each with two offspring. We first analyzed the observed rare-variant genotypes in the family using the kernel test of Chen et al., and then repeated the analysis using our robust within-family association test. As shown in Figure 4, the power of the kernel test using observed genotype information (shown as black bars) is, as expected, more powerful than the same test using within-family information alone (shown in white bars) across different simulation models. In attempts to see whether we could restore some power to the robust test, we then applied our screening procedure to these simulated datasets using between-family information. For each dataset, we tested the between-family components of each of the 10 regions, and then subsequently considered only the top 10%, 20%, 30%, or 40% (based on minimum p-value) of these regions using our within-family test. The results show that, when screening is performed using parental genotype and trait information, our screening procedure restores power to levels similar to those using the observed-genotype information (see top panels of Figure 4). If screening is instead performed using between-family information, the robust within-family association test also shows a power increase, although it is not as notable as using parental information (see bottom panels of Figure 4). Thus, it appears that our initial screening step improves the power of the within-family association test, although preserving appropriate type I error under the null.
, where c ranged from 0.4 to 0.6. We first compared the power of our robust within-family association test to the standard observed-genotype test considered by Chen et al. and Schifano et al. under models with no population stratification (to ensure the power of the observed-genotype test was valid). We generated sequence and trait data on 500 nuclear families each with two offspring. We first analyzed the observed rare-variant genotypes in the family using the kernel test of Chen et al., and then repeated the analysis using our robust within-family association test. As shown in Figure 4, the power of the kernel test using observed genotype information (shown as black bars) is, as expected, more powerful than the same test using within-family information alone (shown in white bars) across different simulation models. In attempts to see whether we could restore some power to the robust test, we then applied our screening procedure to these simulated datasets using between-family information. For each dataset, we tested the between-family components of each of the 10 regions, and then subsequently considered only the top 10%, 20%, 30%, or 40% (based on minimum p-value) of these regions using our within-family test. The results show that, when screening is performed using parental genotype and trait information, our screening procedure restores power to levels similar to those using the observed-genotype information (see top panels of Figure 4). If screening is instead performed using between-family information, the robust within-family association test also shows a power increase, although it is not as notable as using parental information (see bottom panels of Figure 4). Thus, it appears that our initial screening step improves the power of the within-family association test, although preserving appropriate type I error under the null.

 ,
,  , and
, and  . As in Figure 3, for each dataset we used KMFAM to test the observed genotype; then we used our method to test the within-family component without screening, and then applied two screening methods. Top panel: screen by parental genotype. Bottom panel: screen by between-family component. Each result is based on 1,000 replicates.
. As in Figure 3, for each dataset we used KMFAM to test the observed genotype; then we used our method to test the within-family component without screening, and then applied two screening methods. Top panel: screen by parental genotype. Bottom panel: screen by between-family component. Each result is based on 1,000 replicates.Although we obtained our results in Figure 4 under simulation models that assumed no confounding due to population stratification, we also observed similar trends in simulation models that were generated with confounding due to population stratification. Figure 5 presents power results under confounding due to population stratification that assumed a mean trait difference between African and European samples. For the observed-genotype analyses, we report empirical adjusted power accounting for population stratification (black bars) rather than the naïve power that does not account for population stratification (which is invalid). To obtain the empirical adjusted power, we simulated and analyzed null datasets generated with the same amount of confounding as in the power datasets and used the empirical distribution of the null tests to determine an appropriate threshold to declare significance. We then evaluated the observed genotype's power based on this empirical threshold. The remaining bars denote the power of the robust within-family association test, along with variations that screen using parental or between-family information. The results show that screening can improve power of the robust rare-variant test, particularly as the percentage of causal variants and the magnitudes of their effect increase. The results in Figure 5 were for simulated datasets consisting of nuclear families with two offspring each; we saw similar trends when analyzing parent-offspring trios, as well as when a region harbored variants that acted in different directions on outcome (see Supplementary Figures 1 and 2).

 ,
,  , and
, and  . Top panel: screen by parental genotype. Bottom panel: screen by between-family component. Each result is based on 1,000 replicates.
. Top panel: screen by parental genotype. Bottom panel: screen by between-family component. Each result is based on 1,000 replicates.Discussion
In this paper, we proposed a kernel method for analyzing rare-variant sequencing studies in trios and nuclear families that is robust to confounding due to population stratification. We also introduced a screening procedure using parental or between-family information to improve the power of this robust test and showed that this procedure can increase power to levels similar to those of the observed-genotype test when confounding due to stratification is not an issue. In addition to robustness, our approach has many other practical features. The method easily allows for covariates and permits rapid calculation of P-values using analytic procedures. We have implemented our procedure in R software, which is available from our website (see Web Resources). Our approach is computationally efficient, as the analysis of a 30 kb region for 500 nuclear families each of size two takes on average 53.08 seconds on a 768 processor running Linux OS with 2.6 gigahertz of RAM. Based on the computational speed, we believe the approach can be scaled reasonably to whole-exome or whole-genome resequencing studies on a multinode cluster.
Our approach currently considers either parent-offspring trios or nuclear families with an arbitrary number of offspring. In future work, we intend to extend the approach to consider pedigrees of any arbitrary size or structure. Saad and Wijsman [2014] have highlighted the value of using such large families for rare-variant analysis by showing that sequencing a small proportion of subjects although genotyping the remaining subjects using a sparser set of markers, and then subsequently using pedigree information to impute the missing variants among genotyped subjects, will lead to greater power compared to analyzing the sequenced individuals alone. Given such appealing features of such large pedigrees, we will expand our framework to handle such pedigrees by leveraging the work of Abecasis et al. [2000b], which derived within- and between-family information for relatives within general pedigrees. We can then incorporate the within-family information for each subject within our kernel test for inference.
Family-based genetic studies of complex traits occasionally have information available from additional unrelated singletons. Although we cannot use these individuals within our robust within-family association test of rare variation, the information from such singletons can be used in our screening step (treating them in the same way as the parental information) to identify the most interesting regions for follow up using the robust test. Such information could be helpful in screening and should not affect the validity of the second-stage robust test, even if there is confounding due to population stratification and/or coverage differences between the family and unrelated arms of the study.
In this paper, we focused on familial studies of quantitative traits. To extent our method to binary traits, we can look into applying estimating-equation procedures similar to those proposed by Wang et al. [2013] for analyzing observed genotypes. A few additional methods have discussed familial rare-variant analysis of binary traits: Preston and Dudbridge [2014] compared the power of several family-based designs for binary traits, and found that using cases from affected families and unrelated controls often has optimal power. The use of unrelated controls may raise concerns of bias caused by population stratification; to avoid this problem, one could implement an idea similar to Mirea et al. [2012], who adopted a weighting strategy where the between-family and within-family contributions to a test statistic are weighted by a test of population-stratification bias. We will explore these ideas in future work.
Acknowledgments
We thank Cheryl Strauss for her editorial assistance. Dr. Epstein is a paid consultant for Amnion Laboratories.
Web Resources
Cosi simulation package, http://www.broadinstitute.org/∽sfs/cosi
Epstein Software, http://www.genetics.emory.edu/labs/epstein/software
 ,
,  , and
, and  . Left panel: 5% of rare variants in the region are causal. Bottom panel: 15% of rare variants in the region are causal. Each result is based on 1000 replicates.
. Left panel: 5% of rare variants in the region are causal. Bottom panel: 15% of rare variants in the region are causal. Each result is based on 1000 replicates. ,
,  , and
, and  . Left panel: 5% of rare variants in the region are causal. Bottom panel: 15% of rare variants in the region are causal. Each result is based on 1000 replicates.
. Left panel: 5% of rare variants in the region are causal. Bottom panel: 15% of rare variants in the region are causal. Each result is based on 1000 replicates.
