

Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis
Abstract
CO2 electrolysis is a key step in CO2 conversion into fuels and chemicals as a way of mitigating climate change. We report the synthesis and testing of a series of new anion-conductive membranes (tradenamed Sustainion™) for use in CO2 electrolysis. These membranes incorporate the functional character of imidazolium-based ionic liquids as co-catalysts in CO2 reduction into a solid membrane with a styrene backbone. We find that the addition of an imidazolium group onto the styrene side-chains increases the selectivity of the reaction from approximately 25 % to approximately 95 %. The current at 3 V is increased by a factor of 14. So far we have been able to tune these parameters to achieve stable cells that provide current densities higher than 100 mA cm−2 at 3 V cell potential with a CO product selectivity over 98 %. Stable performance was observed for 6 months of continuous operation (>150 000 000 turnovers). These results demonstrate that imidazolium polymers are ideal membranes for CO2 electrolysis.
Introduction
Interest has increased in the conversion of CO2 into fuels and chemicals as a way to mitigate climate change, as a source of renewable feedstocks for fuels and industrial chemicals, and for energy storage.1-20 The electrolysis of CO2 to form CO, formic acid, and other compounds has been observed at industrially relevant rates,21-39 and the processes have high selectivity.40 Thus, electrochemical conversion is a leading candidate for CO2 utilization.
Previously, Aeshala et al.41-44 showed that it is beneficial to optimize the membrane composition to maximize performance for the reaction CO2→CO+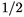 O2. Although the rates observed by Aeshala et al. were below those needed for industrial applications, their work suggested that membrane improvements would be a viable route to improve the performance of electrolyzers for CO2 conversion.
O2. Although the rates observed by Aeshala et al. were below those needed for industrial applications, their work suggested that membrane improvements would be a viable route to improve the performance of electrolyzers for CO2 conversion.
In the work presented here, we wanted to see whether the performance of the CO2 electrolyzer would be increased if we used a membrane that incorporates an imidazolium group. Previously, we discovered that the combination of a metal catalyst such as Ag with a 50 vol % aqueous solution of 1-ethyl-3methylimidazolium tetrafluoroborate (EMIM-BF4) led to a decrease of the overpotential for the reduction of CO2 to CO from approximately 1 V to just 0.17 V.45, 46 The resulting process was not only very energy-efficient but also highly selective, and the conversion to the desired CO product was over 98 %.45, 46 In subsequent work to design a commercially viable electrolyzer for this process, we both broadened and refined the library of candidate ionic liquids.21
One of the difficulties with using ionic liquids is that they are expensive and corrosive. So we wished to find an alternative and we focused on imidazolium-containing polymers. Several imidazolium-containing polymers have been reported previously.47-64 We decided to concentrate on imidazolium-functionalized styrene and vinylbenzyl chloride (VBC) based polymers because they were easy to synthesize and showed the needed performance. These polymers are quite chemically stable and highly conductive. The structure of this polymer is depicted in Figure 1.

General structure of the PSMIM membrane.
With these membranes, we are able to operate CO2 electrolyzers with a membrane electrode assembly (MEA) design using commercially available fuel cell hardware. So far, we have found our membranes with a divinylbenzene (DVB) cross-linking agent to be generally robust and powerful for CO2 reduction.
In this paper we discuss the performance of electrolyzers using these membranes. We also compare their performance to that of eight other membranes that were reported previously for use in CO2 electrolyzers. Our studies indicate that these membranes far outperform any commercially available ion-exchange membranes that can be used for CO2 reduction. We compared our membranes to Nafion, sulfonated poly(ether ether ketone) (SPEEK), polyvinyl alcohol (PVA), polyethylenimine (PEI), Membrane International CMI-7000, Membrane International AMI-7001, phosphoric acid doped polybenzimidazole (PBI), and Neosepta. Each of these materials, if used either as delivered or cast as membranes and implemented in our cells, failed to achieve the kind of activity and selectivity that our membranes show.
Results
A polarization curve measured as CO2 is fed into a water bubbler and subsequently into the cathode flow field at a rate of 20 sccm is shown in Figure 2. The anode was open to the air. Electrochemical analyses such as cyclic voltammetry (CV) were performed by using a potentiostat (PAR VERSTASTAT 3). We start to observe small bumps in the CV between 1.4 and 1.9 V, which shows that there is a small amount of CO2 conversion in this voltage range, but the rate is slow. The CO2 conversion increases rapidly above 1.9 V to reach 140 mA cm−2 at 3 V.

Polarization curve measured with a PSMIM membrane (black) and an AMI-7001 membrane (orange) using identical catalysts and conditions.
We also analyzed the cell output by using GC. At 1.8 V cell voltage, only H2 and CO2 are seen on the cathode, but at 2 V, significant CO production is detected on the cathode and O2 formation is detected on the anode.
The results of the same measurements of an AMI-7001 membrane are shown in Figure 2. The CV curve grows more slowly with none of the bumps in the range of 1.4–1.9 V. The curve only reaches 10 mA cm−2 at 3 V, and H2 is the major product even at 3 V.
Incorporation of an ionomer into cathode
We can further improve the performance of our CO2 electrolyzer by incorporating polystyrene methyl methylimidazolium chloride (PSMIM) or polystyrene tetramethyl methylimidazolium chloride (PSTMIM) ionomer directly into the cathode layer ink. If added in the right amount, this has the effect of maximizing the surface area of the Ag catalyst that is exposed to the ionomer functional groups. The consequences can be quite dramatic, but care must be taken not to add so much ionomer to the ink that CO2 diffusion to catalyst sites is hindered.
A comparison of the cell performance if varying concentrations of PSTMIM ionomer are added to the cathode layer directly is shown in Figure 3. These cyclic voltammograms show that there is little to no difference in the onset cell potential for CO2 reduction if the concentration of ionomer is varied as expected. However, the current density at 3 V changes dramatically with even small adjustments to the ionomer concentration. A change from 1 to 4 wt % more than doubles the current density to a high of approximately 280 mA cm−2. However, further doubling the ionomer concentration to 8 wt % sees the current density decrease precipitously again. At this point, it appears that the blocking of the CO2 diffusion has overwhelmed the benefit from the increased contact between the catalyst metal and co-catalyst ionomer. This blocking effect is seen most dramatically at 32 wt % ionomer at which the current density at 3 V falls close to 40 mA cm−2, which is much lower than peak current obtained if the ionomer is absent from the cathode catalyst layer entirely. As will be explained below, a cell without a cathode layer ionomer can run at 50 mA cm−2 for over 6 months uninterrupted and decrease below 3 V in doing so.

Comparative cyclic voltammograms that demonstrate the effect of the addition of various concentrations of PSTMIM ionomer directly to the cathode catalyst layer. CVs were run in otherwise identical 5 cm2 electrolyzers.
Long-term performance
One of the issues with methylimidazolium-based polymers is that they degrade quickly under alkaline conditions. However, CO2 electrolyzers are not particularly alkaline, so we ran a cell for 6 months at 50 mA cm−2 to determine whether degradation would occur.
The cell performance during a 6 month (4380 h) test is shown in Figure 4. In this case, we ran the cell recycling 10 mM KHCO3 electrolyte solutions through the anode at 2 mL min−1. An IrO2 catalyst was used in the anode. We observed some small leakage of the KHCO3 solution through the gasketing so we needed to refill the anode electrolyte reservoir every 500–1000 h.

Current and voltage measured during a 6 month run at a constant current of 50 mA cm−2 with a catalyst loading of 1 mg cm−2 at room temperature. The sharp drops in voltage are at times when the KHCO3 solution was refreshed.
The initial cell voltage was approximately 3 V. The voltage decreased gradually (i.e., the performance improved) as the run proceeded. The selectivity was almost constant, except for a decrease at the end of the run because of flooding. These data show that the cell and membrane are stable for extended periods and that the catalysts are stable for extended periods.
To put the data displayed in Figure 4 in perspective, a current of 50 mA cm−2 at 95 % selectivity corresponds to a CO production rate of approximately 1.5×1017 molecules s−1 cm−2. The Ag catalyst had a loading of 1 mg cm−2 and a surface area of approximately 10 m2 gm−1, which means that there are approximately 1.5×1016 Ag atoms per cm2 of membrane. That is, the turnover rate is approximately 10 s−1. In 4380 h, the Ag catalyst has undergone 10×4380×3600=158 000 000 total turnovers. This is the largest number of turnovers for CO2 conversion reported to date.
A long-term run was performed with the inclusion of the PSTMIM ionomer into the cathode catalyst layer, and the data for this run are shown in Figure 5.

Current and voltage measured during a 1000 h run. The cell was held at a constant current of 200 mA cm−2 with ionomer incorporated into the cathode and run at room temperature.
Although this cell has not had the opportunity to run for as long as that shown in Figure 4, it has the distinction of operating at a much higher current density, thanks to the embedded ionomer. The cell was held with a steady-state current density of 200 mA cm−2. Notably, this is four times the current density of the cell shown in Figure 4. Here we see that once again, for the duration of 1000 h, the cell is stable enough to operate at or under 3 V even with this very high current density, and a CO selectivity is maintained at above 90 %.
We have also performed experiments using a membrane with a substituted imidazolium polymer in an alkaline water electrolyzer at 60 °C in 1 m KOH.
The change of the cell resistance during these experiments measured by using electrochemical impedance spectroscopy (EIS) is presented in Figure 6. Over the 1000 h run, cell resistance increased at an average of only 4 μΩ h−1, which indicates the high stability of the cell components. Therefore, we predict that the cell voltage and selectivity would remain highly stable well past the 1000 h shown, as a 4 μΩ h−1 increase extrapolates approximately to a four year cell lifetime.

Resistance over time of the alkaline water cell in 1 m KOH at 60 °C. This cell used a membrane with a polysubstituted imidazolium rather than the 1-methylimidazolium shown in Figure 1. Over a 1000 h run, the average rise in resistance was only 4 μΩ h−1.
Elevated temperatures
Both of the cells shown in Figures 4 and 5 were run at room temperature. However, we expect that a consequence of installing these individual units into electrolyzer stacks will be a higher normative operating temperature. A conservative estimate of such a temperature is 50 °C, so we performed some tests to determine the effect of an elevated temperature on the cell output.
An electrolyzer that was heated to 50 °C by using a thermocouple and cartridge heaters that were made for the cell is shown in Figure 7. External tubing was heated to the same temperature by using thermal tape.

Cyclic voltammograms that compare cells of identical structure and composition that operated at room temperature and 50 °C, respectively.
The results show quite clearly that modest heating to 50 °C increases the output current at 3 V by 2–3 times (Figure 7). This is not only because of the presumed increased catalysis of the CO2 reduction reaction but also because of an increase in membrane conductivity with the increasing temperature.
This effect is shown clearly in Figure 8. The cell conductivity was measured across a range of temperatures with two different anolytes for comparison. Notably, the conductivity of these membranes is approximately an order of magnitude higher than that of alkaline membranes reported previously. Unsurprisingly, the overall cell conductivity is higher with a 1 m KOH solution than that with an equally concentrated KHCO3 solution, but the temperature–conductivity trend is identical across the anolytes.

Conductivity data that demonstrate the stable increase in conductivity as a function of thermal uptake up to 80 °C. This cell used a membrane with polysubstituted imidazolium rather than the 1-methylimidazolium shown in Figure 1.
We might consider that upon heating the cell, there is some risk of further encouraging the hydrogen evolution reaction that our catalytic system suppresses. However, our results show that the heated cell maintains 98 % Faradaic efficiency, which obviates any concerns about a loss in product selectivity. This level of modest heating, which we can expect from scaled-up operation, seems to be a clear benefit.
Comparison to other membranes
We also performed experiments to compare the performance of these membranes to that of others reported previously. We used a slightly different procedure to compare to other membranes. We eliminated the KHCO3 solution so as to not poison the acidic cation-exchange membranes and to avoid the leaching of KOH from some of the anion-exchange membranes.
 (1)
(1)
Charts that illustrate the a) total CO current density and b) Faradaic efficiencies of cells that run CO2 electrolysis with a variety of commercial membranes versus our Sustainion membrane. These measurements were collected at 23 °C with 3 V applied cell potential.
The selectivity varies from near 0 to 95 %. The four acidic membranes, Nafion, CMI-7001, SPEEK, and H3PO4-doped PBI show a low selectivity, but interestingly the acidic membrane that contained imidazolium (PBI) shows a higher selectivity than the others.
Alkaline anion-exchange membranes show higher selectivities, which demonstrates that alkaline anion-exchange membranes are preferred, in agreement with the results of Aeshala et al.41-44
It is interesting to compare alkali-doped PSMIM and alkali-doped AMI-7001. The compositions of the two membranes are similar. Both polymers are prepared from a copolymer of styrene and vinylbenzyl chloride, but AMI-7001 is doped with trimethylamine to create a quaternary amine group, whereas PSMIM is doped with imidazole to create an imidazolium. Notably, the presence of the imidazolium increases the selectivity by approximately a factor of 4 and increases the current by a factor of 14. This is consistent with our previous work, which indicates that imidazolium can act as a co-catalyst for the reaction21, 25, 27, 40, 45 and reduce the formation of H2.65
These differences illustrate the importance of the inclusion of an imidazolium in the polymer membranes for the purpose of CO2 electrolysis. This extends not only to CO2 electrolysis for CO production but also to the production of formic acid. Here, we use different metal catalysts (Sn for the cathode side), but again our membrane outperforms commercially available anion-exchange polymers (Figure 10).

Comparison of the current density from a cell that produces formic acid at 3.5 V with our anion-exchange membrane vs. AMI-7001s and ACN (Astom).
PBI versus DVB as a cross-linking agent
One of the issues that we needed to address is whether the divinylbenzene (DVB) that is used as a cross-linking agent in AMI-7001 affected the performance. To explore this possibility, we considered two cross-linking agents: DVB and PBI. We also changed the procedure slightly in that 10 mm KHCO3 electrolyte solutions were recycled through the anode flow field at a rate of 5 mL min−1 as we found that the membrane would dry out if we did not provide water continuously.
The results of these experiments are presented in Table 1. Notably, the cross-linking agents made little difference to the selectivity or rate of the reaction.
Membrane |
Thickness change [%] |
Current density at 3 V [mA cm−2] |
CO selectivity [%] |
|---|---|---|---|
PSMIM |
7.4 |
100±5 |
96.9 |
PSMIM-PBI |
0 |
100±5 |
96.7 |
This all illustrates the important effects of imidazolium polymers on the performance of CO2 electrolyzers. We found that a commercial quaternary-amine-functionalized styrene-vinylbenzyl chloride membrane (AMI-7001) gave a modest performance. However, if we switched to a 1-methylimidazolium-functionalized styrene-vinylbenzyl chloride membrane, the performance was improved greatly. The selectivity went from 25 to 95 % (Figure 9), and the current went from 10 to 140 mA cm−2 (Figure 2). This illustrates the importance of the imidazolium to promote CO2 conversion.
One might wonder whether the difference could be caused by a difference in the conductivity of the membrane. The AMI-7001 membrane is an order of magnitude less conductive than the PSMIM membrane, but the current is more than an order of magnitude less. The voltage drops across all of the membranes shown in Figure 9 are less than 10 mV. So we suggest that the differences in membrane conductivity have little effect on the ability to convert CO2 on the catalyst surface.
Discussion
We can speculate why the imidazolium had these effects. Previously,66 we found that imidazolium groups form a dense, positively charged layer on Ag that repels protons. We attributed the enhanced selectivity seen in ionic liquids to this blocking effect.40, 45, 66 Evidently, we still see this effect even if the imidazolium groups are attached to a polymer. Physically, the polymer swells during the reaction and seems to flow into the spaces in the cathode catalyst layer. Indeed, if we disassemble the cell we find that the catalyst adheres to the membrane strongly. Given the enhanced performance of membranes that swelled more against the cathode layer, it follows logically that an ionomer incorporated directly in the cathode catalyst layer would enhance cell performance even further. The dramatic improvements that we see in Figures 3 and 5 are strong confirmation of this.
Previously, we found that the imidazolium groups decrease the overpotential for the reaction.40, 45 The data here are less clear on that point. We observe some small bumps in the CV at cell potentials between 1.4 and 1.9 V, which suggest that there may be some CO2 conversion, but H2 is the major product. The conversion of CO2 to CO does not really take off until the cell potential is increased to 1.9–2.0 V. This corresponds to a net overpotential (sum of the anode and cathode overpotential) of 0.57–0.67 versus 0.17 V in our previous studies.40, 45 Still, this is a reduced overpotential compared to the results obtained without amines.4
Another interesting feature was that the membrane was stable for 6 months under our normal operating conditions. There was no evidence for membrane degradation over the entire run. Furthermore, overall cell resistance saw a mere 4 μΩ h−1 increase after 1000 h at 60 °C in 1 m KOH. The membrane was also able to perform well if it was heated to 50 °C, and we demonstrated a steady conductivity rise with the increasing temperature up to 80 °C with two different anolytes.
Conclusions
We have developed a membrane for CO2 reduction that far exceeds standards for current density and product selectivity set by commercially available membranes. The key to the potency of this membrane as a helper catalyst is not simply the incorporation of amine groups or doping with strong acids or bases, but the use of the imidazolium cationic functional group. Membranes that incorporate the imidazolium functional group are very powerful co-catalysts for the reduction of CO2, but the creation of such a membrane that is viable in a long-running CO2 electrolyzer requires a careful balance of the mechanical and catalytic properties. These can be tuned by modifying the ratios of the polymer components and the cross-linking agents used.
Experimental Section
Preparation of Sustainion PSMIM: A copolymer of styrene and vinylbenzylchloride functionalized with 1-methyl imidazole
The copolymers of styrene and vinylbenzyl chloride functionalized with 1-methyl imidazole were synthesized following the procedures described by Masel et al.23, 24 First inhibitor-free styrene was prepared by washing styrene (Sigma–Aldrich, St. Louis, MO) with two equal volumes of 7.5 % aqueous NaOH. The inhibitor-free styrene was then washed with four equal volumes of water to make sure it was neutralized and then dried over anhydrous MgSO4. Inhibitor 4-tert-ButylCatechol (TBC) in 4-vinylbenzyl chloride (4-VBC) was removed by extraction with 0.5 % KOH solution until a colorless extract was obtained. This extract was washed with water until neutral and then dried over anhydrous MgSO4. The yield of this step was approximately 70 %.
Poly(4-vinylbenzyl chloride-co-styrene) was synthesized by heating a solution of inhibitor-free styrene (Sigma–Aldrich; 10.0581 g, 96.57 mmol) and 4-vinylbenzyl chloride (Sigma–Aldrich; 6.2323 g, 40.84 mmol) in chlorobenzene (Sigma–Aldrich; 15 mL) at 60–65 °C in an oil bath for 12–18 h under Ar with α,α′-azoisobutyronitrile (AIBN, Sigma–Aldrich; 0.1613 g, 0.99 wt % based on the total monomer weight) as the initiator. The copolymer was precipitated in CH3OH/THF and dried under vacuum to give a polymer yield of approximately 75 %. Larger batches gave yields of approximately 85 %.
The molecular weight of the poly(4-vinylbenzyl chloride-co-styrene) samples were measured by using gel permeation chromatography (GPC). Different runs gave average molecular weights of 47 000–51 000 atomic units (A.U.) with a polydispersity index (PDI) of 1.4–1.5.
PSMIM was synthesized by adding 1-methylimidazole (Sigma–Aldrich; 2.8650 g, 0.0349 mol), which is an alkylimidazolium, to a solution of poly(4-VBC-co-St) (5.0034 g) in anhydrous DMF (Sigma–Aldrich; 30 mL). The mixture was then stirred at RT for 0.5–1 h and then heated at 110–120 °C for 50.3 h to form a PSMMIM solution with greater than 95 % functionalization.
The membranes were prepared by the solution-casting and evaporation method. The polymers were dissolved in DMF to form a 15–35 wt % solution. The polymer solutions were then cast onto glass slides and dried in a vacuum oven at 80 °C for 1 h then 150 °C for 1 h to produce membranes that were approximately 60 μm thick. The dried membranes were peeled off from the glass slides by soaking the glass slides in 1 m KOH at RT. The membranes were then soaked in 1 m KOH for at least 12 h to exchange the chloride ions with hydroxide ions. Finally, the obtained membranes were rinsed thoroughly with deionized water before further use. Details of the procedures are given in our patents.23, 24
At the end of the procedure, the membranes are solid with no clear pores. We pressurized the anodes with water at 5 psi and did not detect water leakage after 24 h. There are of course nanopores in the membrane.
Preparation of other polymers that were used for comparison
Nafion 117 was purchased from Ion Power Technologies, Inc. in Wilmington, DE. Neosepta BP-1E was purchased from Ameridia Division of Eurodia Industrie S.A. in Somerset, NJ. CMI-7000 and AMI-7001 were purchased from Membranes International Inc. in Ringwood, NJ. PVA (341584) was purchased from Sigma–Aldrich Corporation in St. Louis, MO. PEI (408727) was purchased from Sigma–Aldrich Corporation in St. Louis, MO. A PEEK film was purchased from CS Hyde Company in Lake Villa, IL. PBI was purchased from PBI Performance Products, Inc. (Rock Hill, SC).
PVA and PVA/PEI
The membranes were prepared according to the procedure described by Aeshala et al.41, 42 Either PVA (9 g) alone or a mixture of PVA (6 g) and PEI (3 g) were dissolved in deionized water at 90 °C. Dissolved solutions were then cast in petri dishes to produce membranes that were approximately 60 μm thick. After the cast films had dried, they were immersed in glutadehyde (10 % solution in acetone) and mixed with small volumes of catalytic HCl to encourage cross-linking. The films were rinsed several times with deionized water and soaked in 0.5 m KOH for at least 24 h before use in a cell.
AMI and CMI
AMI-7001 from Membranes International is a copolymer of styrene and vinylbenzyl chloride that has been functionalized by trimethyl amine and cross-linked with divinylbenzene (DVB). This is different to the Fumatech product, which is based on polypropylene. The as-delivered AMI membranes were treated as described by Aeshala et al.[43] First they were immersed in 0.5 m KOH for at least 24 h, then subsequently soaked in deionized water to remove excess KOH. A similar process was used for CMI, except that 0.5 m H2SO4 was used in place of KOH, as the CMI membrane is a cation-exchange membrane.
Neosepta BP-1E
As recommended by the manufacturer, this membrane was pretreated solely by soaking it briefly in deionized water before it was assembled into the cell.
Nafion 117
The Nafion membrane was prepared according to a standard activation procedure. First, it was boiled for 1 h each in a 5 % solution of H2O2, Millipore water (18.2 MΩ cm−1), 0.5 m solution of H2SO4, and Millipore water successively. It was then assembled into a cell.
Phosphoric acid doped PBI
A 50 μm thick PBI membrane was acid-doped by soaking it in 0.5 m phosphoric acid for 24 h. It was then soaked in deionized water for 1 h to remove excess acid then assembled into a cell and tested.
SPEEK
As-delivered PEEK film (1 g) was immersed in concentrated sulfuric acid (50 mL) under constant agitation for 50 h. At this point, all of the PEEK had dissolved and was sulfonated fully to SPEEK. Next, Millipore water (200 mL) was allowed to cool to near 0 °C in an ice bath. The SPEEK solution was poured slowly into this cold water under constant agitation. SPEEK precipitated out of solution, was filtered, and was then washed several times with Millipore water to remove excess sulfuric acid. It was then dried at 100 °C for 8 h in a vacuum oven. Next, this SPEEK precipitate was dissolved in dimethylacetamide, and the resulting membrane solution was cast on a glass slide to produce membranes that were approximately 60 μm thick. After the membrane had dried, it was assembled into a cell and tested.
Electrochemical cell assembly and operation
Electrodes were prepared by creating nanoparticle inks and applying them to porous substrates. The cathode ink was made by adding Ag nanoparticles (30 mg) to distilled water (0.1 mL) and isopropanol (0.2 mL). This mixture was sonicated for 1 min then spray-painted onto a 2.5 cm×2.5 cm square cut out of carbon gas diffusion layer (Sigracet 35 BC GDL, Ion Power). IrO2 and RuO2 anodes were prepared in the same way but with the addition of 5 % PTFE (100 μL) solution as binder. The anode catalyst was then sintered for 1 h at 330 °C. In some cases, PSMIM or PSTMIM ionomer was added to the cathode ink. In these cases, the painted cathodes were dried in an oven at 80 °C for 50 min, 120 °C for 50 min, and then allowed to soak in 1 m KOH for 1 h to encourage ion exchange.
Electrochemical testing cells were assembled by sandwiching a membrane between a Ag cathode and IrO2 anode such that both electrode catalyst layers were facing the membrane. Each catalyst was surrounded by a layer of gasketing for protection and electrical insulation, and this assembly was then mounted into Fuel Cell Technologies 5 cm2 fuel cell hardware with serpentine flow channels.
Next CO2 humidified at 50 °C was supplied to the cathode, and the cell was set to 3 V. In most experiments, the anode was open to the air (no anolyte), and the only flow into the cathode was humidified CO2 (no catholyte). An exception experiment presented in Figure 4 in which 10 mm KHCO3 electrolyte solutions were passed through the anode at 2 mL min−1. Again, there was no catholyte. The cathode output composition was analyzed by using an Agilent 6890 GC with a thermal conductivity detector (TCD; Agilent Technologies, Santa Clara, CA) equipped with a Carboxen 1010 P LOT GC column (30 m×320 μm; Sigma–Aldrich). No heating was applied to the cell.
Acknowledgements
Parts of this work were supported by ARPA-E under contract DE-AR0000345 and by 3M. The opinions here are those of the authors and may not reflect the opinions of ARPA-E or 3M. Assistance from colleagues, collaborators, and friends from 3M is gratefully acknowledged. The authors have received U.S. Patents 9,370,773 and 9,481,939 and submitted U.S. Patent applications number US 2016/0251766 and 15/158,227, which claim the membranes and electrolyzer designs discussed here. Dioxide Materials is planning to offer improved versions of these membranes for sale. The authors all have a financial stake in the outcome of this sale.

