

Conformations within soluble oligomers and insoluble aggregates revealed by resonance energy transfer
Abstract
A fluorescently labeled 20-residue polyglutamic acid (polyE) peptide 20 amino acid length polyglutamic acid (E20) was used to study structural changes which occur in E20 as it co-aggregates with other unlabeled polyE peptides. Resonance energy transfer (RET) was performed using an o-aminobenzamide donor at the N-terminus and 3-nitrotyrosine acceptor at the C-terminus of E20. PolyE aggregates were not defined as amyloid, as they were nonfibrillar and did not bind congo red. Circular dichroism measurements indicate that polyE aggregation involves a transition from α-helical monomers to aggregated β-sheets. Soluble oligomers are also produced along with aggregates in the reaction, as determined through size exclusion chromatography. Time-resolved and steady-state RET measurements reveal four dominant E20 conformations: (1) a partially collapsed conformation (24 Å donor–acceptor distance) in monomers, (2) an extended conformation in soluble oligomers (>29 Å donor–acceptor distance), (3) a minor partially collapsed conformation (22 Å donor-acceptor distance) in aggregates, and (4) a major highly collapsed conformation (13 Å donor–acceptor distance) in aggregates. These findings demonstrate the use of RET as a means of determining angstrom-level structural details of soluble oligomer and aggregated states of proteins. © 2009 Wiley Periodicals, Inc. Biopolymers 93: 299–317, 2010.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at [email protected]
INTRODUCTION
While numerous studies have explored fundamental elements (mechanisms, intermediate structures, and theoretical determinants) of protein folding, these fundamental elements of protein aggregation remain largely unexplored and unknown.1-11 Although some advances have been made, routine structural determination of high molecular-weight soluble oligomers and insoluble aggregates remains a challenge.8-11 Many theoretical models of protein assembly are predicated on the idea that the same computational parameters used to describe protein folding are also equally appropriate to characterize protein aggregation.12-18 While this assumption may be valid, the lack of structures of soluble oligomers and insoluble aggregates make it difficult to compare the accuracy of structural predictions from any given theoretical model. Given the role protein oligomers and aggregates play in both healthy biological processes and disease mechanisms, the lack of structural understanding about protein oligomers and aggregates will continue to remain a gap in scientific knowledge which hinders advances in technological and medical advances.8-11
A number of fundamental questions remain unanswered about the structure and biological activity of protein oligomers and aggregates. For example, do protein chain conformations differ in oligomers/aggregates of different shapes and sizes? Also, do toxic oligomers/aggregates differ from nontoxic oligomers/aggregates level of polypeptide conformations? Until the structures of peptides and proteins in oligomer and aggregated states can both be determined, these questions will remain unanswered.
Some progress has been made towards obtaining the molecular structures of protein aggregates. Solid-state NMR can determine peptide structures in amyloid fibers.8-10 X-ray crystallography can be performed if amyloid microcrystals can be produced from small (eight residues of less) amyloid peptides.11 While these two methods perform well on the final equilibrium products of aggregation reactions, neither approach can easily determine the structure of transiently populated aggregation intermediates nor follow an aggregation reaction in real time. Also, the development of new techniques are required to study protein conformations within soluble oligomer species, believed to be a likely cause of neurodegenerative diseases such as Alzheimer's.19, 20 The present study aims to demonstrate the applicability of using resonance energy transfer (RET) to study protein conformations in both insoluble protein aggregates and soluble oligomers alike.
Each RET experiment provides a measurement of the distance between a fluorescent donor (D) and acceptor group (A) attached to a molecule.7, 21-32 Since fluorescence can be measured directly on protein samples in real-time, RET is a useful technique to determine structural properties of “difficult” and transiently populated protein states. RET has been used in numerous studies to study conformations of monomeric proteins in membranes and natively disordered states.7, 21-27, 29-31 However, RET has not been applied as universally to study protein conformations in aggregates or soluble oligomers.28, 32-35 A handful of steady-state RET-based studies have provided valuable structural information on aggregated conformations of a transthyretin peptide, huntingtin exon 1, and α-synuclein, although such studies only capture ensemble-averaged RET efficiencies in a given sample.28, 33, 35 Also, a RET study conducted on Aβ11–25 aggregates indicates that a time-resolved RET approach may offer additional insights into the structure of protein aggregates beyond the steady-state approach.32
The present study combined steady-state and time-resolved RET approaches on a well-studied polyglutamic acid (polyE) peptide aggregation model using a specific polyE peptide length (E20) as a RET structural probe.7, 36-41 In this study, the self-consistency of both steady-state and time-resolved RET was assessed as the peptides transitioned from monomers into aggregates and soluble oligomers. In addition, direct measurements relevant to the Förster distance calculation, i.e., fluorescence anisotropy (κ2), donor/acceptor spectral overlap (J), and donor quantum yield (ϕD), were assessed to estimate the overall precision of the RET method on aggregated states.
PolyE aggregation was used in the present study instead of more disease-related amyloid peptides due to the wealth of background studies of polyE and relative ease of transitioning polyE from soluble monomer states into insoluble aggregates. For over 40 years, polyE has been used as a model for protein folding, owing to a pH-dependent helix-coil transition.7, 36-41 However, polyE also forms aggregates below pH 4.5 making it a convenient model for protein aggregation by pH jump. The advantages of aggregation studies using polyE are (1) initial monomeric polyE reactant states are easily produced at pH >5 and (2) the reaction rate can be controlled by the final pH of the aggregation solution (minutes–weeks).42-44 Thus, polyE aggregation is a highly practical model of aggregation to test RET measurements.
Recently, RET has successfully characterized monomeric conformations of E20 throughout a pH-dependent helix-coil transition.7 The present study is an extension of this previous study to E20 conformations adopted in aggregates and soluble oligomers. Starting from 100% monomer states, the presence of discrete soluble oligomer and aggregate populations with unique RET efficiencies were tracked throughout the entire course of aggregation. While measurements on sample mixtures of monomers, oligomers, and large aggregates were conducted, isolated aggregates were purified from soluble species by centrifugation and soluble oligomers were isolated from monomers through size exclusion chromatography (SEC). Ultimately, these D–A distances will be used to model structures of E20 aggregates and soluble oligomers with theoretical methods.13, 14, 45-47 Using a similar approach, future RET studies with amyloid peptides can be used to facilitate theoretical–experimental investigations into disease-related protein aggregates.7, 27
MATERIALS AND METHODS
Sample Preparation and Buffer Conditions
Two peptides were synthesized for the present study: (1) ABZ-E20-Y (D-E20) and (2) ABZ-E20-YNO2 (D-E20-A). In these peptides, ABZ denotes an N-terminal o-aminobenzamide (RET donor), Y denotes a C-terminal tyrosine, and YNO2 denotes a C-terminal 3-nitrotyrosine (RET acceptor).7, 23, 25, 48, 49 All peptides were obtained through custom synthesis (Anaspec) with purity >95% was achieved with High Performance Liquid Chromatography (HPLC) purification and confirmed through matrix-assisted laser desorption/ionization (MALDI) mass spectrometry and molecular weight analysis on a Beckman XLA analytical centrifuge. Unlabeled polyE peptides were polydisperse, with sizes ranging between 20 ± 15 residues (Sigma P1943).
Aggregation Experiments
The present study uses a modified protocol of Fandrich et al.50 to aggregate polyE peptides and is shown in Figure 1. Initial monomers are obtained from phosphate-buffered pH 6 stock solutions of 250 μM RET labeled peptide (D-E20 or D-E20-A) and 4 mg/ml (∼1 mM) unlabeled polyE. To initiate aggregation, 1 mg/ml (∼250 μM) unlabeled polyE is brought to pH 4.1 in 100 mM sodium phosphate and 100 mM NaCl. The solution is immediately centrifuged at 12,000 rpm and filtered to remove any initial aggregates. Under these conditions, polyE aggregates will nucleate within approximately 2 days and fully reach equilibrium between all monomer, oligomer, and aggregate species after 2 weeks (Figure 1A). For RET studies, the following samples were studied, containing polyE plus the indicated fluorescent RET probe: (1) 2.5 μM D-E20 (donor only), (2) 10 μM D-E20 (donor only), (3) 2.5 μM D-E20-A (donor + acceptor), (4) 10 μM D-E20-A (donor + acceptor), (5) 10 μM free ABZ, and (6) polyE alone (no RET probe). To determine error estimates, two completely independent experiments consisting of Samples 1, 3, 5, and 6 and polyE alone and three completely independent experiments consisting of Samples 2 and 4 were performed. Analysis of the centrifuged pellet and SEC fractions of the centrifuged supernatant were performed on time points from single experiments of Samples 2 and 4.

Schematic of the aggregation RET experiment. RET labeled peptides (D-peptide-A) are aggregated in an excess of unlabeled peptide (shown by “A”) producing an ensemble of monomers, soluble oligomers, and aggregates (center EM image). At various days of the experiment, species within this mixed sample are separated by centrifugation into the pellet containing the aggregates (shown by “B”) and supernatant containing monomers and soluble oligomers (shown by “C”). On some days of analysis, the monomers and soluble oligomers in the centrifuged fraction are isolated by size exclusion chromatography (shown as “D”). On days of analysis, RET measurements are conducted on uncentrifuged samples (a), aggregates in the resuspended pellet (b), centrifuged supernatant (c), and SEC fractions (d).
On each day of analysis during each experiment (days 0, 1, 2, 3, 5, 7, 9, and 11), two aliquots from the sample were removed. The first aliquot was analyzed without any further treatment and addresses structures of the complete ensemble of monomer, oligomer, and aggregate populations which existed at that time point (Figure 1a). The second aliquot was centrifuged at 12,000 rpm to separate insoluble aggregates in the pellet (Figure 1B) from soluble monomers/oligomers in the supernatant (Figure 1C). The aggregate-containing pellet was buffer-washed 3× through repeated resuspension and recentrifugation and was finally resuspended and analyzed (Figure 1b). The monomers and oligomers in the supernatant were similarly analyzed as a mixture (Figure 1c) and after further purification by SEC (Figure 1d). Analyses indicated in Figure 1a–1d include steady-state fluorescence emission and anisotropy, time-resolved fluorescence and anisotropy decays, light scattering, turbidity, thioflavin T (ThT) binding, and congo red (CR) binding. Additionally, electron microscopy was performed on the final day (day 11) of single experiments on uncentrifuged (Figure 1A/a), centrifuged pellet (Figure 1B/b), and centrifuged supernatant (Figure 1C/c) for samples with D-E20 (Sample 2), D-E20-A (Sample 4), and polyE only (Sample 6). Finally, circular dichroism (CD) spectra were acquired for uncentrifuged (Figure 1A/a) and centrifuged supernatant (Figure 1C/c) polyE only (Sample 6) on days 0 and 11.
Electron Microscopy
Electron microscopy was conducted using a Morgagni 268 transmission electron microscope (TEM) equipped with a Hamamatsu digital camera. A 70-μl aliquot of each sample was placed on a formar grid and let sit for 20 min. The sample was fixed for 10 min with one drop of 1% glutaraldehyde in 100 mM phosphate buffer. The sample was rinsed using doubly distilled water and followed by staining with 1% uranyl acetate for 10 min. The sample was rinsed again with water and allowed to dry. Approximately 10 scans within the field were collected. Scans shown in the present study are representative of the majority of oligomer and aggregate mesoscopic structures observed within the field.
Steady-State Spectroscopy
 (1)
(1)
CD spectra of 250 μM polyE prior to aggregation on day 0 (), the uncentrifuged ensemble of monomers/oligomers/aggregates on day 11 (--------), the centrifuged supernatant containing monomers/oligomers on day 11 (………), and the concentration-normalized centrifuged supernatant sample on day 11 (-•-•-•).
In Eq. (1), Ivv is fluorescence collected with a vertical excitation and vertical emission polarizer position, Ivh, is the fluorescence collected with a vertical excitation and horizontal emission polarizer position. The G-factor, G, is the ratio of fluorescence measured with polarizer orientations Ihv/Ihh. In cases where significant background light scattering or turbidity was present in fluorescence or absorbance measurements, the background light scattering from the matched polyE-only control sample was subtracted from the sample.
Aggregation and Amyloid Properties
Aggregation was quantitated in the samples through measurements of turbidity and light scattering. Turbidity was measured as 600 nm absorbance using a Cary 100 ultraviolet/visible (UV/Vis) absorbance spectrophotometer and collected similar to absorbance measurements at 600 nm. Light scattering was measured as fluorescence using a PTI Quanta Master spectrophotometer with both excitation and emission at 600 nm.
 (2)
(2) (3a)
(3a) (3b)
(3b) (4)
(4) (5)
(5)Size Exclusion Chromatography
Size exclusion chromatography separations were performed on a 120 ml volume GE Life Sciences Sephacryl S-200HR column with an optimal separation range between molecular sizes of Mr 5000–250,000, connected to a Pharmacia P-1 peristaltic pump and Gilson fraction collector. Calibration of this column system with dextran blue and cytochrome c reveal that V0 is 40 ml and Vt is 120 ml. All SEC separation procedures were performed on samples after centrifugation and removal of large aggregates, which would not elute through the column and would clog the column matrix. The volume of sample applied was 1 ml, the flow rate was 0.5 ml/min and samples were collected from the eluent in 3 ml volumes. The amount of free ABZ, D-E20, or D-E20-A in each column fraction was assessed using fluorescence at 320 nm excitation and 420 nm emission (PTI Quanta Master).
Time-Resolved Fluorescence
Fluorescence lifetimes were measured on D-E20, D-E20-A, and free ABZ using a PTI EasyLife equipped with a 295 nm LED. Sample preparation prior to acquisition followed previously described methodology.7, 24 ABZ emission was selected by a 395 nm cut-on filter. Prior to and after each measurement, the ABZ steady-state fluorescence spectra were acquired to ensure that minimal (<5%) photobleaching had occurred.
 (6)
(6)In Eq. (6), the fluorescence lifetime τi and signal amplitude Ai of each exponential kinetic process i, the number of kinetic processes N, and the final signal value A(∞) at equilibrium were assessed using the reduced chi-squared (χ ) statistic, randomness in the fit residuals, and the logical consistency of the fitted parameters.52
) statistic, randomness in the fit residuals, and the logical consistency of the fitted parameters.52
To determine the time-resolved fluorescence anisotropy decay, 0° and 90° polarized luminescence decays, Ivv(t) and Ivh(t), were collected separately with a sheet polarizer in the emission light path. On the instrument used, a G-factor of ∼1 was determined through the ratio Ivv(t)/Ivh(t) of 2.5 μM ABZ, which rotates quickly enough to emit an equal fluorescence signal in either polarization orientation. The deconvoluted 0° and 90° polarized luminescence decays, Ivv(t) and Ivh(t), were fit globally using Savuka software to a single exponential fit [Eq. (6)] of Eq. (1), and the corresponding amplitudes used in the global fit to determine the initial anisotropy amplitude r0 and rotational correlation time τc of the slower anisotropy decay phase.53 It should be noted that, although both a fast (τc ∼ ps) and slow (τc ≥ ns) decay are present in time resolved anisotropy measurements of D-E20, only the slower (τc ≥ ns) decay is measurable with the PTI EasyLife instrument due to signal-to-noise limitations.23 As in previous studies, the fast decay (τc ∼ ps) from the fundamental anisotropy of ABZ (rf ∼ 0.35) to the initial amplitude of the slower decay (r0) is not observed and only the time constant of the slow decay (τc ≥ ns) from r0 to the baseline can be fit from the data.7, 25
Evaluation of the Förster Distance
 (7)
(7) (8)
(8)The integral in Eq. (8) is evaluated numerically across the total overlap range of 300–600 nm shown in Supporting Information Figure S1 [shown as the right side of Eq. (8)]. FD(λ) is the intensity at each wavelength in the fluorescence spectrum of D-E20, normalized so the total area under the fluorescence curve is one. εA(λ) is the molar extinction coefficient of the nitrotyrosine acceptor, expressed in M−1 cm−1, at each wavelength (in nm) in the summation. Under most conditions used in the present study (100 mM phosphate, 100 mM NaCl, pH 4), the D-E20-A Förster distance R0 was determined to be 20 Å for monomeric D-E20-A.
RET Analysis
For steady-state RET measurements, the average RET efficiency E was calculated from the fluorescence intensity of the D-E20-A peptide (IDA) and fluorescence intensity of the D-E20 peptide (ID) at equivalent concentrations and in the absence of light scattering and inner-filter effects. In samples which are turbid from the presence of aggregates, conversion from the observed values of I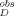 and I
and I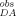 to the actual values of ID and IDA requires Eqs. 9a and 9b. The equations shown in 9a and 9b are for the D-E20 peptide but are the same as was used for the D-E20-A peptide.
to the actual values of ID and IDA requires Eqs. 9a and 9b. The equations shown in 9a and 9b are for the D-E20 peptide but are the same as was used for the D-E20-A peptide.
 , is calculated by subtracting the background light scattering, measured as observed fluorescence intensity, I
, is calculated by subtracting the background light scattering, measured as observed fluorescence intensity, I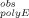 , in the polyE-only control (Sample 6 in “Aggregation Experiments” mentioned above), acquired on the same day of analysis as I
, in the polyE-only control (Sample 6 in “Aggregation Experiments” mentioned above), acquired on the same day of analysis as I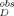 .
.
$$ I_{\rm D}^{\rm bc}=I_{\rm D}^{\rm obs}-I_{\rm polyE}^{\rm obs}\,$$.
(9a)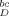 from Eq. (9a) for inner-filter effects resulting from high turbidity in a sample, where large aggregates scatter incident excitation light and emitted photons from fluorescence emission.55
from Eq. (9a) for inner-filter effects resulting from high turbidity in a sample, where large aggregates scatter incident excitation light and emitted photons from fluorescence emission.55
 (9b)
(9b)In Eq. (9b), A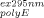 is the apparent absorbance at the excitation wavelength (295 nm for the present study) and A
is the apparent absorbance at the excitation wavelength (295 nm for the present study) and A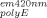 is the apparent absorbance at the primary emission wavelength (420 nm for the present study) in the polyE-only control (Sample 6 in “Aggregation Experiments” mentioned above), acquired on the same day of analysis as I
is the apparent absorbance at the primary emission wavelength (420 nm for the present study) in the polyE-only control (Sample 6 in “Aggregation Experiments” mentioned above), acquired on the same day of analysis as I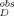 . Assuming that A
. Assuming that A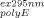 and A
and A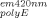 were measured in a 1-cm cuvette, parameters ℓex is the fluorescence cuvette pathlength (cm) in the excitation direction and ℓem is the fluorescence cuvette pathlength (cm) in the emission direction.
were measured in a 1-cm cuvette, parameters ℓex is the fluorescence cuvette pathlength (cm) in the excitation direction and ℓem is the fluorescence cuvette pathlength (cm) in the emission direction.
 (10a)
(10a) (10b)
(10b) (11)
(11)It is important to note that E determined using steady-state RET [Eq. (10a)] reflects the average E of a mixture of monomers, oligomers, and aggregates which exist simultaneously in a sample while E determined using fluorescence lifetimes [Eq. (10b)] reflects E from subpopulations within the mixture of monomers, oligomers, and aggregates with unique lifetimes τDA and RET properties.
RESULTS AND DISCUSSION
Aggregates of PolyE are not Amyloid
Previous work has suggested that polyE aggregates might be amyloid as a fibrillar morphology was observed in these studies.50 However, polyE aggregates in the present study are not fibrillar and instead appear to have an amorphous/dendritic morphology (Figure 1, center EM image). These EM structures were demonstrated to result from polyE aggregates as they were removed through centrifugation (Figure 1, lower right EM image) but retained in the pellet (Figure 1, upper right EM image). It should be noted that the centrifuged sample (Figure 1, lower right EM image) contains worm-like structures which could reflect the mesoscopic structures of soluble oligomers. While buffer-only control EM studies do not show these worm-like structures (not shown), it is still not clear if such structures are indeed that of soluble oligomers or are instead (a) a small fraction of residual aggregates remaining after centrifugation, (b) nascent aggregates nucleated from monomers or oligomers in the short time (∼minutes) between sample centrifugation and TEM grid deposition, or (c) unavoidable polyE aggregation formed from polyE peptides resulting from the process of fixing and drying each TEM sample. Thus, these TEM studies convincingly demonstrate large amorphous/dendritic structures of polyE aggregates but whether worm-like polyE soluble oligomers exist is still under study.
To better characterize the aggregates, soluble oligomers, and monomers in samples, the amyloid properties of turbidity, light scattering, ThT, and CR dye binding were conducted. These tests are shown in Supporting Information Figure S2 for uncentrifuged and resuspended pellet samples containing large aggregates and Figure S3 for centrifuged samples containing soluble monomers/oligomers only. For uncentrifuged and resuspended pellet samples in Figure S2, measurements of turbidity, light scattering, and ThT binding all detect large aggregates which begin forming after a 2-day lag phase and reach a maximum after 7 days (Figures S2A–C). For centrifuged samples in Figure S3, the turbidity, light scattering, and ThT properties are close to background levels (Figures S3A–C) indicating that the entirety of turbidity, light scattering, and ThT binding in Figures S2A–C can be attributed to the aggregates removed by centrifugation. However, CR binding, a traditional amyloid indicator, is shown to bind neither the aggregates (Figure S2D) nor the species in the centrifuged sample (Figure S3D). In Figures S2 and S3, similar results are obtained regardless of whether the polyE sample contains D-E20 (open circles), D-E20-A (filled gray circles), ABZ (squares), or nothing added (triangles), indicating that RET probes do not appear to perturb the polyE aggregation reaction.
While ThT binding is an amyloid property (Figure S2C), ThT binding is not sufficient to classify polyE aggregates as amyloid in the present study as the polyE aggregates are not fibrillar (Figure 1, center) and do not bind CR (Figure S2D). In addition, ThT binding to nonamyloid aggregates has been noted in previous studies, demonstrating that ThT binding is a necessary but not sufficient amyloid classification test.56, 57 A study by Fandrich et al.50 reported fiber-like structures of aggregated polyE, although ThT and CR binding was not conducted. While the fibrillar polyE aggregate morphology reported in the Fandrich et al. study contrasts with the amorphous morphology observed in the present study, this discrepancy could be explained by the use of different polyE chain molecular weights (74 kDa for Fandrich et al. study, 3 kDa in the present study). In either case, amyloid or nonamyloid, polyE is still an excellent model system to assess the use of RET to study the general process of protein aggregation and soluble oligomer formation.
PolyE monomers and oligomers in the centrifuged samples also lack amyloid properties given the complete absence of ThT and CR binding (Figures S3C and S3D). It should be noted that small levels of turbidity and light scattering are present in Figures S3A and S3B at an approximately ∼1% that of aggregates in Figures S2A and S2B. However, in further SEC studies of soluble polyE/E20 species, no column fractions produced light scattering levels above baseline (data not shown), indicating that this slightly elevated turbidity and light scattering in centrifuged samples is likely due to a small portion (<1%) of polyE aggregates remaining in the sample after centrifugation. For RET studies, this minor aggregate population is not significant and does not alter fluorescence-based measurements of RET in the centrifuged sample fractions.
Apparent β-Sheet Content Increases in PolyE Aggregates but not Soluble Oligomers
Previous CD, Fourier transform infrared spectroscopy (FTIR), and small-angle X-ray scattering (SAXS) studies have supported a partial (∼60%) α-helical structure of polyE at pH 4.7, 37-40 In the present study, a pre-aggregated CD spectrum, acquired at the initiation of the aggregation experiment, displays two spectral minima at 208 and 222 nm with magnitudes consistent with a 60% α-helical peptide (Figure 2, solid line). After 11 days of aggregation, a second CD spectrum with a minimum near 226 nm was found to be consistent with previous aggregated polyE CD spectra (Figure 2, dashed line).58 Aggregated polyE spectra are consistent with a red-shifted β-sheet spectrum due to “line-flattening” effects from light scattering.58 While both previous and present aggregated polyE CD spectra indicate β-sheet content in polyE peptides, these CD spectra do not clearly reveal the type of β-sheet conformations (i.e., parallel, anti-parallel, β-turn, fully extended, etc.).
The CD spectrum of the centrifuged sample on day 11 (Figure 2, dotted line), consisting of only the remaining soluble monomer and oligomer species, generally appears similar to the initial monomer spectrum on day 0 but at a reduced mean residue ellipticity (deg cm2 dmol–1 residue–1) due to the loss of aggregated peptide during centrifugation. Assuming an approximate 30% remaining soluble polyE at day 11 (see Figure 3A), scaling this spectrum by 3.3 produces a normalized CD spectrum of the centrifuged sample (Figure 2, dot-dashed line). While this normalized day 11 centrifuged spectrum does appear similar to the day 0 α-helical CD spectrum, slight differences are found with a more negative and blue shifted band near 208 nm and a less negative band at 222 nm. Given that an approximate 1:2 monomer:oligomer ratio determined from SEC (see Figure 4), this spectrum can be considered largely representative of polyE soluble oligomers. Thus, while some α-helix content may remain, soluble oligomers exhibit an increase in the characteristic random coil spectral component and a decrease in the α-helical component.

Measurements of (A) fraction soluble polyE (FSP), (B) fraction soluble ABZ (FSA), and (C) fraction of initial fluorescence in samples of aggregating 250 μM polyE in the presence of 2.5 μM D-E20 (•), 2.5 μM D-E20-A (○) 10 μM D-E20 ( ), 10 μM D-E20-A (○), 2.5 μM free ABZ (□), or polyE only (▵). For 10 μM data, the error bars represent standard deviation from three independent experiments. All other error bars show the high/low range of two independent experiments.
), 10 μM D-E20-A (○), 2.5 μM free ABZ (□), or polyE only (▵). For 10 μM data, the error bars represent standard deviation from three independent experiments. All other error bars show the high/low range of two independent experiments.

Size exclusion chromatography plots of centrifuged sample fractions of D-E20 on day 0 (——), D-E20 on day 7 (--------), D-E20 on day 11 (………), free ABZ on day 0 ( ), free ABZ on day 11 (
), free ABZ on day 11 ( ). SEC column fractions corresponding to monomer (m), soluble oligomer (o), and free ABZ (a) are indicated above the three dominant elution peaks.
). SEC column fractions corresponding to monomer (m), soluble oligomer (o), and free ABZ (a) are indicated above the three dominant elution peaks.
Fluorescent ABZ-Labeled E20 Peptides Co-Aggregate with Unlabeled PolyE Peptides but Free ABZ does not
Figures 3A–3C demonstrate that D-E20 and D-E20-A co-aggregate with polyE but free ABZ does not. Figure 3A shows the loss of soluble polyE (and E20) peptides calculated by Eq. (4) and indicates that the rate of decreasing soluble polyE (a) mirrors the aggregation kinetics in Figures S2A, S2B, and S2C and (b) is not changed by the presence of added D-E20, D-E20-A, and free ABZ. Figure 4B shows the loss of soluble ABZ calculated by Eq. (5) for the ABZ group in D-E20, ABZ donor group in D-E20-A, and free ABZ. Figure 3B shows that D-E20 and D-E20-A co-aggregate with polyE at a rate similar to polyE aggregation in Figure 3A while the free ABZ is not co-aggregated after centrifugation. Similar results are obtained using the fraction of initial fluorescence after centrifugation in Figure 3C, where the decrease in D-E20 and D-E20-A fluorescence intensity mirrors the decrease in soluble polyE and ABZ as measured with absorbance in Figures 3A and 3B, respectively. The rate and extent of fluorescence decrease in Figure 3C is similar, within error, between D-E20 and D-E20-A peptides and at either concentration used (2.5 or 10 μM). In additional control experiments, the decrease of D-E20 and D-E20-A within 11 days, as measured after centrifugation by absorbance in Figure 3B and by fluorescence in Figure 3C, only occurs in the presence of 250 μM polyE, as 10 μM D-E20 and D-E20-A aggregate considerably more slowly (weeks) in the absence of polyE at pH 4.1 (data not shown). Thus, the best explanation for these results is that D-E20 and D-E20-A co-aggregate uniformally throughout the polyE chains. Also, the constant level of free ABZ absorbance in Figure 3B and fluorescence in Figure 3C, despite extensive polyE aggregation, demonstrates that ABZ does not itself participate in the aggregation process and is therefore a good choice for a RET donor.
D-E20 Quantum Yield Remains Similar between Oligomers and Monomers
Figure 4 shows the results of SEC conducted on centrifuged sample fractions of aggregating polyE on days 0, 7, 11 with D-E20 added and days 0, 11 for free ABZ added, using fluorescence of the ABZ as a sensitive probe for the presence of different soluble oligomer states. In Figure 4, it is clear that the monomer population, denoted by the label “m” above fractions 22–24, decreases during the aggregation reaction to approximately 10% of the initial value by day 11 (black lines). One notable observation is that a second oligomer state, denoted by label “o” above fractions 28–30, increases to a fraction of 20% of the initial fluorescence on day 11 (dotted black line). Similar SEC profiles to D-E20 are also found with the analogous experiment of aggregating polyE but with D-E20-A added (data not shown). To test whether residual aggregates after centrifugation might be affecting the SEC column performance and resulting in the appearance of these two SEC peaks, controls using free ABZ in the presence of polyE monomers on day 0 and in the presence of centrifuged polyE aggregates on day 11 were conducted and show a single peak labeled “a” above fractions 38–40 under both conditions (Figure 4, gray lines). The single free ABZ peak on days 0 and 11 demonstrates that the two peaks for D-E20 and D-E20-A on days 7 and 11 are the result of two distinct soluble states, monomer and oligomer, and not due to the altered performance of the SEC column on day 11.
While the presence of two distinct peaks for monomer and oligomer is confirmed in Figure 4, it is interestingly that the oligomer peak would elute later than the monomer when one would expect them to elute earlier if they were larger. The exact physical reasons for this are not known. Possible explanations may be that soluble oligomers interact with the stationary phase of the column, which would slow their progress through the column relative to the monomers. Also, if the oligomers are small enough (dimers, trimers, etc.) and happen to be considerably more conformationally flexible than monomers, they may be better able to adopt extended states which may better occupy the pores within the column matrix than the monomers.
Regardless of the mechanism, SEC separates monomers from oligomers and permits RET analysis of D-E20-A in the purified monomer and oligomer states. Also, knowing that the monomer:oligomer ratio is 1:2 permits calculation of the quantum yield (ΦD) of D-E20 in the oligomer state. Given that the ΦD of the D-E20 monomer was determined to be 0.26, the similarity between the value of the final fraction of initial absorbance of D-E20 in Figure 3B (0.26) and final fraction of initial fluorescence of D-E20 in Figure 3C on day 11 (0.28) indicates that the average ΦD of the D-E20 in the monomer:oligomer ensemble is also ∼0.26. As Figure 4 shows that oligomers populate >50% of the monomer: oligomer ensemble on day 11, it can be concluded that the ΦD of the D-E20 in soluble oligomers is also 0.26.
D-E20 Quantum Yield Remains Similar between Aggregates and Monomers
Figure 5A shows the steady-state fluorescence spectra of 2.5 μM D-E20 and D-E20-A on the initial day (day 0) and at the end (day 11) of the polyE aggregation reaction. On day 0, the fluorescence of D-E20-A is approximately 70% of D-E20, consistent with previous RET studies on monomeric D-E20 and D-E20-A.7 Free ABZ fluorescence at 2.5 μM on day 0 was normalized to a 0.98 fraction of D-E20 to aid the viewer in viewing both ABZ and D-E20 spectra, which would overlap completely if normalized equally. After aggregation on day 11, the apparent fluorescence of D-E20, D-E20-A, and free ABZ decrease from their values on day 0, demonstrating that the presence of polyE aggregates alters the apparent level of fluorescence in D-E20, D-E20-A, and free ABZ. While the decrease in D-E20-A fluorescence can be attributed to RET, the reason for the decrease in D-E20 and ABZ is less clear.

(A) Normalized steady-state fluorescence spectra and (B) normalized steady-state fluorescence spectra additionally corrected for light scattering and inner filter effects [Eq. (9a)/(9b)] of 250 μM polyE samples containing 2.5 μM of D-E20 on day 0 (——), D-E20 on day 11 (), D-E20-A on day 0 (--------), D-E20-A on day 11 ( ), ABZ on day 0 (………) and ABZ on day 11 (). All D-E20 and D-E20-A samples are normalized by the same factor such that the maximum value of D-E20 on day 0 is 1.0. All ABZ samples are normalized by the same factor such that the maximum value of ABZ on day 0 is 0.98 (to avoid overlap with D-E20 spectra).
), ABZ on day 0 (………) and ABZ on day 11 (). All D-E20 and D-E20-A samples are normalized by the same factor such that the maximum value of D-E20 on day 0 is 1.0. All ABZ samples are normalized by the same factor such that the maximum value of ABZ on day 0 is 0.98 (to avoid overlap with D-E20 spectra).
Two reasons exist to explain the lower D-E20 and ABZ fluorescence in aggregates in Figure 5A. First, D-E20, D-E20-A, and ABZ may all co-aggregate with polyE after which the ABZ group is quenched in the aggregated state by the molecular environment of the polyE aggregates. Second, the apparent ABZ fluorescence may be reduced by inner-filter effects, i.e., scattering of excitation and emission light by polyE aggregates, not donor quenching.
The present study strongly supports the presence of inner filter effects, not quenching of ABZ, as the reason for the decrease in fluorescence of free ABZ and D-E20 between uncentrifuged samples on days 0 and 11. First, it is clear from Figure 5A that free ABZ fluorescence drops at a very similar fraction (∼0.5) as that of D-E20 between day 0 and day 11. However, examination of centrifuged samples of aggregating polyE containing free ABZ in Figures 3B and 3C show that ABZ absorbance and fluorescence is virtually identical on day 0 to all subsequent days. Thus, free ABZ clearly does not bind polyE aggregates yet demonstrates the same reduction in fluorescence as D-E20 in the presence of polyE aggregates in Figure 5A, a reduction which can only be attributed to inner-filter effects. Second, based on the assumption that aggregates produce both an increase in apparent fluorescence due to light scattering and a decrease in apparent fluorescence due to inner-filter effects, Eqs. (9a) and (9b) are used to correct the spectra in Figure 5A to the spectra and these corrected spectra shown in Figure 5B. It is clear from Figure 5B that free ABZ and D-E20 spectra on day 11 are corrected very close to the initial spectra on day 0, again supporting the idea that inner-filter effects, not donor quenching, are responsible for decreasing D-E20 fluorescence in the aggregated state.
As no donor quenching occurs between D-E20 in the monomer and aggregated states, it can be concluded that the ΦD of the D-E20 in soluble oligomers is 0.26, as with the monomer. While the corrected fluorescence of D-E20 is similar on days 0 and 11 in Figure 5B, it is notable that the corrected D-E20-A spectra on day 11 in Figure 5B is considerably lower (∼ 0.25 fraction of initial) than that of D-E20-A on day 0. This decrease is the result of RET, which is increased for D-E20-A peptides in the aggregated state.
Steady-State RET Measures Compact E20 Conformations in Aggregates
Figure 6A shows the corrected steady-state fluorescence (ID, IDA, or IABZ) in aggregating polyE samples containing 2.5 μM D-E20 (filled diamonds), 10 μM D-E20 (filled gray circles), 2.5 μM D-E20-A (open diamonds), 10 μM D-E20-A (open circles), 2.5 μM ABZ (squares), or polyE only/nothing added (triangles). The data shown in Figure 6A is the result of the correction of observed fluorescence data through Eqs. (9a) and (9b) and is shown for 2.5 μM data in Figure 5B for days 0 and 11. In Figure 6A, the fluorescence of both 2.5 μM D-E20 and D-E20-A are normalized by the same factor such that the fluorescence of 2.5 μM D-E20 on day 0 are 1.0. Similarly, the fluorescence of both 10 μM D-E20 and D-E20-A are normalized by the same factor such that the fluorescence of 10 μM D-E20 on day 0 are 1.0. The fluorescence of 2.5 μM ABZ data is normalized such that the fluorescence of 2.5 μM ABZ on day 0 is 1.0. Finally, the background light scattering in the polyE-only sample is normalized by the same factor as 10 μM D-E20, but is not corrected with Eq. (9a)/(9b), and is shown for reference. All samples in Figure 6A were uncentrifuged and the indicated fluorescence emission includes contributions from a mixture of monomers, soluble oligomers, and aggregates.

(A) Steady-state fluorescence, normalized and corrected for light scattering and inner filter effects [Eq. (9a)/(9b)], of uncentrifuged samples of aggregating 250 μM polyE with 2.5 μM D-E20 (•), 10 μM D-E20 (), 2.5 μM D-E20-A (○), 10 μM D-E20-A (○), and 2.5 μM free ABZ (□). PolyE only samples (▵) were normalized but not corrected for light scattering and inner-filter effects. All 2.5 μM D-E20 and D-E20-A samples are normalized by the same factor such that area under the 2.5 μM D-E20 spectrum on day 0 is 1.0. All 10 μM D-E20, D-E20-A, and polyE only samples are normalized by the same factor such that area under the 10 μM D-E20 spectrum on day 0 is 1.0. All 2.5 μM free ABZ samples are normalized by the same factor such that area under the 2.5 μM free ABZ spectrum on day 0 is 1.0. (B) RET efficiency determined from steady-state fluorescence in uncentrifuged samples of aggregating polyE with 2.5 μM D-E20/D-E20-A (•), aggregating polyE with 10 μM D-E20/D-E20-A (), and 10 μM D-E20/D-E20-A in the absence of polyE (▵). For 10 μM data, error bars represent standard deviation from three independent experiments. All other error bars show the high/low range of two independent experiments.
In Figure 6A, the corrected fluorescence of 2.5 μM D-E20, 10 μM D-E20, and 2.5 μM ABZ remain constant, within the errors of the experiment. However, the corrected fluorescence of D-E20-A decreases significantly during aggregation, indicating that RET increases in aggregated D-E20-A. In Figure 5A, no difference is found between the fluorescence of either D-E20 at 2.5 and 10 μM or between D-E20-A at 2.5 and 10 μM, demonstrating that all RET occurs between D-A on the same D-E20-A chain and no RET occurs between D and A on neighboring chains. For reference, the background light scattering at 420 nm is shown for polyE-only samples with no ABZ present (triangles) and is demonstrated to be reasonably low even in the presence of extensive aggregation due to the large (100 nm) Stokes shift of ABZ fluorescence. This minimal background on day 11 is approximately 4% of the 10 μM D-E20 fluorescence (shown in Figure 6A) and 16% of the 2.5 μM D-E20 fluorescence (not shown). Regardless of the level of this background scattering, it is correctly accounted for in all steady-state RET calculations through Eq. (9a).
Figure 6B shows the RET efficiency E calculated from steady-state fluorescence of 2.5 μM D-E20/D-E20-A (filled diamonds) and 10 μM D-E20/D-E20-A (open circles), all in the presence of 250 μM polyE and in uncentrifuged samples. The value of E in D-E20-A increases from 0.25 as a monomer to 0.67 after aggregation. Similar results between 2.5 and 10 μM samples demonstrate that the E is due entirely to intramolecular RET between D and A on the same D-E20-A chain. As a control, the value of E in monomeric 10 μM D-E20/D-E20-A (no polyE added) is shown to remain constant at ∼ 0.24 over the same time course (triangles) as expected as this sample remains monomeric for approximately 2 weeks (data not shown).
The results in Figures 6A and 6B demonstrate that the average D-E20-A RET efficiency E increases as D-E20-A assembles into polyE aggregates. In this aggregated state, the D and A ends of D-E20-A will be much closer than in the monomer state. If one assumes that a highly extended structure would have a large end-to-end distance and a compact, or collapsed, structure would place the ends close together, then the ends of D-E20-A are consistent with a compact structure. It should be noted that RET calculations from steady-state fluorescence measurements on centrifuged pellet (Figure 1B/b), centrifuged supernatant (Figure 1C/c), and SEC fractions (Figure 1D/d) were not performed as there was some uncertainty in the ratio of D-E20-A and D-E20-A concentrations after centrifugation. This ratio must be constant to compare E between different samples and the ability to accurately assess this ratio in the pellet, supernatant, and SEC fractions was limited. However, at a qualitative level, it is notable that the fluorescence of D-E20-A in the pellet fraction is approximately 15% of D-E20 (E ∼ 0.85), further supporting the idea that aggregates contain compact D-E20-A conformations.
Time-Resolved RET Identifies Highly Compact E20 Conformations in Aggregates and Extended E20 Conformations in Soluble Oligomers
While RET calculations from steady-state fluorescence reveal the average RET efficiency E in a sample, time-resolved fluorescence can reveal the number of subpopulations and the specific RET E of each subpopulation in a sample. Furthermore, time-resolved fluorescence does not explicitly require equal D-E20 and D-E20-A concentrations to accurately measure RET E. Thus, time-resolved fluorescence is an ideal approach to determine RET E in all sample fractions (uncentrifuged, centrifuged pellet, centrifuged supernatant, and SEC fractions).
Table I shows the results of one exponential and two exponential fits [Eq. (6)] of aggregating polyE samples with free ABZ, D-E20, or D-E20-A added in different sample fractionation conditions (uncentrifuged, centrifuged pellet, centrifuged supernatant, and SEC fractions) at the beginning (day 0) and end (day 11) of the polyE aggregation reaction. ABZ fits present the average and high/low data error range of two repeated experiments with 2.5 μM ABZ. D-E20 fits present the average and standard deviation of two repeated experiments with 2.5 μM D-E20 and three repeated experiments with 10 μM D-E20. D-E20-A fits present the average and standard deviation of two repeated experiments with 2.5 μM D-E20-A and three repeated experiments with 10 μM D-E20-A.
| Sample | SEC Fractions | Day | Fraction A1 | τ1 (ns) | Fraction A2 | τ2 (ns) | χ2r (1 exp) | χ2r (2 exp) |
|---|---|---|---|---|---|---|---|---|
| ABZa | ||||||||
| U | — | 0 | 1.0 (0.0) | 6.2 (0.6) | — | — | 0.78 (0.13) | 0.79 (0.13) |
| U | — | 11 | 1.0 (0.0) | 6.2 (0.4) | — | — | 0.38 (0.05) | 0.39 (0.05) |
| P | — | 0 | — | — | — | — | — | — |
| P | — | 11 | — | — | — | — | — | — |
| C | — | 0 | 1.0 (0.0) | 6.3 (0.5) | — | — | 0.74 (0.18) | 0.74 (0.18) |
| C | — | 11 | 1.0 (0.0) | 6.2 (0.5) | — | — | 0.39 (0.06) | 0.37 (0.09) |
| C | 38–40b | 11 | 1.0 (0.0) | 6.1 (0.3) | — | — | 0.30 (0.02) | 0.29 (0.01) |
| D-E20c | ||||||||
| U | — | 0 | 1.0 (0.0) | 6.1 (0.04) | — | — | 0.51 (0.29) | 0.51 (0.30) |
| U | — | 11 | 1.0 (0.0) | 6.3 (0.5) | — | — | 0.24 (0.06) | 0.22 (0.06) |
| P | — | 0 | — | — | — | — | — | — |
| Pd | — | 11 | 1.0 (0.0) | 5.9 (0.2) | — | — | 0.44 (0.16) | 0.34 (0.16) |
| C | — | 0 | 1.0 (0.0) | 6.0 (0.1) | — | — | 0.62 (0.27) | 0.62 (0.27) |
| C | — | 11 | 1.0 (0.0) | 5.5 (0.9) | — | — | 0.33 (0.15) | 0.33 (0.16) |
| C | 22–24b | 0 | 1.0 (0.0) | 6.0 (0.1) | — | — | 0.33 (0.04) | 0.29 (0.07) |
| C | 22–24b | 11 | 1.0 (0.0) | 6.4 (1.9) | — | — | 0.34 (0.18) | 0.33 (0.19) |
| C | 28–30b | 11 | 1.0 (0.0) | 5.6 (0.4) | — | — | 0.30 (0.08) | 0.29 (0.15) |
| D-E20-Ac | ||||||||
| U | — | 0 | 1.0 (0.0) | 4.7 (0.03) | — | — | 0.57 (0.27) | 0.56 (0.29) |
| U | — | 11 | 0.41 (0.09) | 4.5 (0.9) | 0.59 (0.09) | 0.59 (0.16) | 0.62 (0.10) | 0.25 (0.10) |
| P | — | 0 | — | — | — | — | — | — |
| Pc | — | 11 | 0.31 (0.03) | 3.9 (0.2) | 0.69 (0.03) | 0.48 (0.08) | 1.17 (0.18) | 0.29 (0.02) |
| C | — | 0 | 1.0 (0.0) | 4.7 (0.1) | — | — | 0.63 (0.30) | 0.56 (0.32) |
| C | — | 11 | 1.0 (0.0) | 5.1 (0.3) | — | — | 0.31 (0.08) | 0.26 (0.06) |
| C | 22–24b | 0 | 1.0 (0.0) | 4.8 (0.1) | — | — | 0.26 (0.14) | 0.23 (0.05) |
| C | 22–24b | 11 | 1.0 (0.0) | 5.0 (0.3) | — | — | 0.31 (0.15) | 0.29 (0.04) |
| C | 28–30b | 11 | 1.0 (0.0) | 5.6 (0.3) | — | — | 0.28 (0.06) | 0.26 (0.14) |
- a Errors show the high/low range of two independent experiments (shown in parentheses).
- b Standard deviation obtained using the three SEC fractions (single experiment).
- c Unless noted otherwise, standard deviations obtained using five independent experiments (shown in parentheses).
- d Standard deviation obtained using the final 4 days (5, 7, 9, and 11) of a single experiment.
In Table I, fits of free ABZ fluorescence decays using Eq. (6) reveal that only a single exponential decay with τ1 ∼ 6.2 ns is needed to adequately minimize the χ parameter in all sample fractionation conditions. The pellet was not fit since ABZ did not co-aggregate with polyE and remained soluble under all conditions (Figures 3B and 3C). A 2 exponential fit does not yield any improvement in χ
parameter in all sample fractionation conditions. The pellet was not fit since ABZ did not co-aggregate with polyE and remained soluble under all conditions (Figures 3B and 3C). A 2 exponential fit does not yield any improvement in χ under any conditions. The single exponential fit of the ABZ fluorescence decay not new and is consistent with previous time-resolved fluorescence studies of ABZ.7, 25, 48 However, what is novel about this finding is that time-resolved fluorescence is capable of accurately measuring the fluorescence lifetime of the ABZ decay even in complex samples, such as extensive polyE aggregation. This feature is crucial for the accurate analysis of D-E20 and D-E20-A lifetimes in the presence of aggregates.
under any conditions. The single exponential fit of the ABZ fluorescence decay not new and is consistent with previous time-resolved fluorescence studies of ABZ.7, 25, 48 However, what is novel about this finding is that time-resolved fluorescence is capable of accurately measuring the fluorescence lifetime of the ABZ decay even in complex samples, such as extensive polyE aggregation. This feature is crucial for the accurate analysis of D-E20 and D-E20-A lifetimes in the presence of aggregates.
Table I also shows that, in general, fits of D-E20 fluorescence decays using Eq. (6) reveal that only a single exponential decay with τ1 ∼ 6.1 ns is needed to adequately minimize the χ parameter of the fit to the fluorescence decays in nearly all sample fractionation conditions. The possible exception is found with centrifuged (C) samples and oligomer-eluting SEC fractions 28–30 on day 11, which still fit well to a single exponential decay but with a slightly lower τ1 of ∼5.6 ns. This suggests that the D-E20 lifetime in oligomers may be slightly less than in monomers or aggregates, although the difference is not outside the statistical error in this parameter (±0.4). In either case, the rate of fluorescence decay of D-E20 remains largely unchanged under conditions where monomers, soluble oligomers, and aggregated states are selectively enriched.
parameter of the fit to the fluorescence decays in nearly all sample fractionation conditions. The possible exception is found with centrifuged (C) samples and oligomer-eluting SEC fractions 28–30 on day 11, which still fit well to a single exponential decay but with a slightly lower τ1 of ∼5.6 ns. This suggests that the D-E20 lifetime in oligomers may be slightly less than in monomers or aggregates, although the difference is not outside the statistical error in this parameter (±0.4). In either case, the rate of fluorescence decay of D-E20 remains largely unchanged under conditions where monomers, soluble oligomers, and aggregated states are selectively enriched.
In contrast to ABZ and D-E20, fits of D-E20-A fluorescence decays differ significantly in both the number of exponentials and the optimal parameters through which the best fit is achieved. Samples in which the monomer is the predominant species (uncentrifuged U and centrifuged C samples on day 0, monomer-eluting SEC fractions 22–24) produce D-E20-A fluorescence decays which fit best to a single exponential with τ1 ∼ 4.7 ns. D-E20-A fluorescence decays in samples where aggregates predominate (uncentrifuged U and pellet P on day 11) fit best to two exponentials. However, the parameters of the two exponential fit differ from U and the P samples on day 11. Both produce a dominant (∼60–70% amplitude) phase with a short time constant (τ2 ∼ 0.5) and a lower amplitude (30–40%) phase with a longer time constant The longer D-E20-A time constant of U on day 11 (τ1 ∼ 4.5) is similar to that of the monomer samples while the longer time constant of P on day 11 (τ1 ∼ 3.9) is slightly less than that of the monomer samples. It is somewhat surprising that the P sample would produce a 30% amplitude phase with a long time constant (τ1 ∼ 3.9) as one might assume that all D-E20-A peptides within the aggregates are compact, in which case a single exponential 0.5 ns decay time constant would be expected. At present, it is not clear whether the slower decay phase (τ1 ∼ 3.9) represents 30% of D-E20-A conformations within the aggregates or if this is due to resolubilized monomeric/oligomeric D-E20-A which quickly re-equilibrated from the pellet at 0.30 fractional population during the brief pellet resuspension process.
Table I also reveals that soluble oligomers have a different D-E20-A fluorescence decay time constant than monomers. Samples where more than 50% of the D-E20-A molecules are in soluble oligomers [centrifuged (C) samples and SEC fractions 28–30 on day 11], produce fluorescence decays which fit to a single exponential but with an increased time constant (τ1 ∼ 5.1 ns) relative to monomer samples(τ1 ∼ 4.7 ns). For SEC fractions 28–30 containing pure soluble oligomers, this time constant is τ1 ∼ 5.6 ns while, in centrifuged samples (C), the time constant is τ1 ∼ 5.1 ns. Presumably, the centrifuged sample time constant (τ1 ∼ 5.1 ns) is not as high as SEC purified oligomers in fractions 28–30 (τ1 ∼ 5.6 ns) due to the presence of 30–40% monomers (τ1 ∼ 4.7 ns), which could not be resolved as a second exponential phase and were therefore averaged into the single exponential fit. It is also somewhat surprising that the longer time constant from uncentrifuged sample (U) on day 11 (τ1 ∼ 4.5 ns) did not match that of the single time constant fitted from the centrifuged sample (C) on day 11 (τ1 ∼ 5.1 ns) if indeed they are both fitting the same population of D-E20-A in soluble monomers/oligomers. As a possible explanation, the fraction of D-E20-A in soluble oligomers of uncentrifuged samples on day 11 is relatively low (∼20%) versus aggregates and monomers which may hinder accurate fitting. This possibility is supported by the relatively high standard deviation (±0.9) observed in fitting of the longer τ1 ∼ 4.5 ns phase in uncentrifuged samples on day 11. Thus, there is no statistical difference between the τ1 ∼ 4.5 ns phase in uncentrifuged samples and the τ1 ∼ 5.1 ns phase in centrifuged samples on day 11.
Table II shows the RET efficiencies, calculated from Eq. (10b), for subpopulations of D-E20-A molecules in different sample fractionation conditions. For exclusively monomeric D-E20-A conformations in uncentrifuged U and centrifuged C samples on day 0 and monomer-eluting SEC fractions 22–24, a RET efficiency of ∼0.25 is measured, consistent with previous studies.7 For the uncentrifuged sample on day 11 containing monomers, oligomers, and aggregates, two D-E20-A subpopulations are determined with RET efficiencies E1 = 0.24 and E2 = 0.91, although the error of E1 is quite high (± 0.16). For the resuspended pellet on day 11 consisting of presumably 100% aggregates, two D-E20-A subpopulations are determined with RET efficiencies E1 = 0.33 and E2 = 0.92, although it is unclear at present if the E1 population is actually a component of the aggregate. For centrifuged C samples on day 11, a single subpopulation is determined with a low RET efficiency E1 = 0.06 despite the presence of both monomers and oligomers in the sample. Isolation of the soluble oligomer fraction in SEC fractions 28–30 reveal that the soluble oligomer RET efficiency E1 = 0.00 is indeed quite low.
| E20 Sample | SEC Fractions | Day | E1 | E2 |
|---|---|---|---|---|
| U | — | 0 | 0.25 (0.03) | — |
| U | — | 11 | 0.24 (0.16) | 0.91 (0.02) |
| P | — | 0 | — | — |
| Pb | — | 11 | 0.33 (0.04) | 0.92 (0.01) |
| C | — | 0 | 0.24 (0.05) | — |
| C | — | 11 | 0.06 (0.06) | — |
| C | 22–24c | 0 | 0.20 (0.03) | — |
| C | 22–24c | 11 | 0.22 (0.04) | — |
| C | 28–30c | 11 | 0.00 (0.12) | — |
- a Unless noted otherwise, standard deviations obtained using five independent experiments (shown in parentheses).
- b Standard deviation obtained using the final 4 days (5, 7, 9, and 11) of a single experiment.
- c Standard deviation obtained using the three SEC fractions (single experiment).
From the results in Table II, at least two distinct conformational populations of D-E20-A in each sample are observed, one with a lower RET efficiency E1 and the other with a high RET efficiency E2. In general, it appears that E1 in uncentrifuged samples (U), centrifuged samples (C), or SEC fractions derives from soluble D-E20-A species, i.e., monomers and/or oligomers,, where E1 decreases when the fraction of soluble oligomers increases. Despite the clear identification of both monomers and oligomers in centrifuged samples (C) on day 11 (see Figure 4), a two exponential fit could not identify the distinct, albeit similar, D-E20-A fluorescence decay times of the monomer and oligomers in Table II. Thus, the RET efficiency of the centrifuged sample on day 11 is simply observed as a single RET efficiency with an averaged value (E1 = 0.06) between monomers (E1 ∼ 0.25) and soluble oligomers (E1 ∼ 0.00), whose actual values were determined after purification by SEC. For the E1 = 0.33 subpopulation in resuspended pellet (P) samples, it is still currently unknown if this particular E1 = 0.33 population is soluble or insoluble or whether it is monomeric, oligomeric, or aggregate. Further studies into E1 conformational states within the resuspended pellet is the subject of current work.
Although the nature of the E1 species may vary depending on the sample studied, E2 is clearly associated with D-E20-A conformations in aggregates. This statement is supported by the observation that, in uncentrifuged samples (U), the increase in fractional amplitude of the τ2 fluorescence phase (A2) mirrors quite well the general aggregate properties in Figures S2A–S2C and loss of soluble peptide in Figures 3A–3C throughout the time-course of the reaction (Figure 7A).

(A) The fraction amplitude A2 from the fast phase of fitted D-E20-A fluorescence decay obtained from uncentrifuged aggregating 250 μM polyE samples with 2.5 μM D-E20-A (•) and 10 μM D-E20-A (). (B) The amplitude weighted RET efficiency 〈E〉 obtained from time resolved fluorescence decays from 2.5 μM D-E20/D-E20-A (•) and 10 μM D-E20/D-E20-A () and the steady-state RET efficiency determined from 2.5 μM D-E20/D-E20-A (▵) and 10 μM D-E20/D-E20-A (▵). For 10 μM data, error bars represent standard deviation from three independent experiments. All other error bars show the high/low range of two independent experiments.
 (12)
(12)In Eq. (12), A1 reflects the corresponding value of A1 in Table I, A2 reflects the corresponding value of A2 in Table I, E1 reflects the corresponding value of E1 in Table II, and E2 reflects the corresponding value of E2 in Table II for uncentrifuged samples of D-E20-A on days 0, 1, 2, 3, 5, 7, 9, and 11. The comparison of 〈E〉 from time-resolved fluorescence and E from steady-state fluorescence is shown in Figure 7B and shows good overall agreement at both 2.5 or 10 μM peptide concentrations. Thus, RET efficiencies determined using both steady-state and time-resolved fluorescence are self-consistent in the present study.
In a small peptide such as D-E20-A, one can use D-A RET efficiency as a variable correlated with peptide chain compactness, From these results, one can therefore conclude that monomeric D-E20-A peptide chains are moderately compact (E = 0.25) and D-E20-A peptides in soluble oligomers are more extended and less compact (E = 0.00). For D-E20-A peptides in aggregates, the majority are found to be highly compact (E = 0.92), in agreement with steady-state measurements of RET. However, a minor (∼30%) subpopulation may exist within the aggregates with a RET efficiency closer to that of monomers (E = 0.33).
E20 Peptides are Flexible in Monomers and Soluble Oligomers but More Rigid in Aggregates
Both steady-state (Figures 8A and 8B) and time-resolved (Figures 8C and 8D) fluorescence anisotropy measurements were conducted to investigate fluorophore rigidity of D-E20 during assembly into soluble oligomers and aggregates. Figure 8A indicates that aggregation of D-E20-A is accompanied by an increase in steady-state anisotropy to a limiting value of 〈r〉 ∼ 0.24 by day 11. Also in Figure 8A, the consistent value 〈r〉 ∼ 0 for free ABZ in the presence of increasing levels of polyE aggregates over days 0–11 further confirms that polyE light scattering is effectively removed by background subtraction (see “Steady-State Spectroscopy” in Materials and Methods) and that free ABZ does not interact with polyE aggregates (also demonstrated in Figures 3B and 3C). Also shown in Figure 8A is the value of 〈r〉 measured from resuspended pellet (Figure 1B/b) samples which show a value of 〈r〉 ∼ 0.31. Figure 11B indicates that centrifuged samples have low steady-state values of 〈r〉 ∼ 0.02, indicating that monomers and soluble oligomers have low 〈r〉 while the higher values of 〈r〉 shown in Figure 8A are due entirely to fluorophore rigidity in the aggregate population of D-E20-A.

(A and B) The steady-state fluorescence anisotropy 〈r〉 of (A) uncentrifuged and (B) centrifuged samples of aggregating 250 μM polyE containing 2.5 μM D-E20 (•), 10 μM D-E20-A (○), 2.5 μM free ABZ (□), and 10 μM D-E20 in a resuspended pellet (▵). (C and D) The initial time-resolved fluorescence anisotropy r0 of (C) uncentrifuged and (D) centrifuged samples of aggregating polyE containing 2.5 μM D-E20 (•), 10 μM D-E20-A (○), 2.5 μM free ABZ (□), and 10 μM D-E20 in a resuspended pellet (▵). For 10 μM data, error bars represent standard deviation from three independent experiments. All other error bars show the high/low range of two independent experiments.
To provide information needed determine the error in RET measurements, time-resolved anisotropy (r0) measurements were also conducted on uncentrifuged (Figure 8C) and centrifuged samples (Figure 8D) using global fitting (see “Time-Resolved Fluorescence” in Materials and Methods). In the case of uncentrifuged samples (Figure 8C), fits on days 0–2 produced similar results with a initial anisotropy of r0 ∼ 0.05 and a rotational correlation time τc of approximately 2 ns (data not shown), consistent with a previous study on monomeric D-E20.7 Between days 3 and 11, the value of r0 increased to 0.18, consistent with steady-state anisotropy measurements in Figure 8A. Values of r0 ∼ 0.26 calculated for resuspended pellet samples provide a direct measure of the initial anisotropy r0 for D-E20 peptides in the aggregated state. On days 3–11 and in all resuspended pellet samples, the rotational correlation time τc fit to very large values (>107 ns) (data not shown), indicating that the measurable anisotropy component of D-E20-A in aggregates decays much slower than the lifetime of the fluorophore (∼ 6 ns). Also in Figure 8C, the constant value of r0 ∼ 0 in control measurements of free ABZ in the presence of increasing levels of polyE aggregates over days 0–11 confirms that polyE light scattering does not introduce significant error to measurements of r0 in these studies, and again shows that ABZ does not interact with polyE aggregates (also demonstrated in Figures 3B, 3C, and 8C). The low constant value of r0 ∼ 0.5 of all centrifuged D-E20 samples in Figure 8D mirrors the low constant value of 〈r〉 ∼ 0.02 for all centrifuged D-E20 samples in Figure 8B, further supporting highly flexible donor in monomers and soluble oligomers. The value of τc from time resolved fits of centrifuged samples shown in Figure 8D is similar to monomer samples (∼2 ns).
Thus, both steady-state and time-resolved anisotropy measurements indicate that the ABZ donor fluorophore of D-E20 remains flexible (r0 = 0.05, τc ∼ 2 ns) in monomer and soluble oligomer states while being substantially more rigid in the aggregated polyE environment. For purposes of RET, it is important to determine a discrete value of the larger value of r0 for D-E20 in the aggregated state, which is provided by a direct measurement of r0 ∼ 0.26 in the resuspended pellet.23, 59 This value is consistent with the slightly lower value obtained from uncentrifuged samples on day 11 (r0 ∼ 0.18), which is a mixture of ∼30% monomer (r0 ∼ 0.05) and ∼70% aggregated D-E20 (r0 ∼0.26) peptides. While these results highlight the increased rigidity of the ABZ donor in aggregated D-E20 peptides (r0 ∼ 0.26), there is clearly some residual flexibility of D-E20 in the aggregated state as this value is less than the fundamental r0 of ABZ, estimated to be ∼0.35.48
Förster Distance Considerations
A summary of the relevant RET parameters of D-E20 and D-E20-A in monomers, soluble oligomers, and large aggregates is shown in Table III. For calculation of the D–A distance RDA, it is important to consider the intrinsic error and/or variation in the Förster distance R0. Intrinsic error of R0 increases when the donor or acceptor are rigid and is determined from fluorescence anisotropy, where a high value of r0 indicates a deviation of κ2 from the standard value of 2/3 in Eq. (8). While it is very difficult to know the true value of κ2 when r0 > 0, an appropriate theoretical treatment can determine the range of possible values of κ2 for a given value of r0.23, 59 Using this approach, the intrinsic error in κ2 and κ2-related variables (RDA and R0) are shown in parentheses in Table III. Such intrinsic error should be considered separate from and additive with the experimental error inherent in the study (shown in all data figures). There is a higher intrinsic error in κ2, RDA, and R0 for aggregated D-E20-A (r0 = 0.26) than for monomers and soluble oligomers (r0 = 0.05). Nonetheless, this intrinsic error in aggregated RDA (±3 Å) cannot account for the large measured difference of RDA between aggregated and monomeric D-E20-A (11 Å difference) or aggregated and oligomeric D-E20-A (>16 Å difference).
| E20 Species | |||
|---|---|---|---|
| Physical property | Monomers | Soluble oligomers | Aggregate |
| Time-resolved r0 | 0.05 | 0.05 | 0.26 |
| Rotational correlation time τc (ns) | 2.0 | 2.0 | >107 |
| κ2 | 2/3 (0.6–0.9) | 2/3 (0.6–0.9) | 2/3 (0.3–2.4) |
| D-E20 quantum yield | 0.26 | 0.26 | 0.26 |
| J (M−1 cm−1 nm4 × 1015) | 1.2 | 1.2 | 1.2b |
| Förster distance R0 in D-E20-A (Å) | 20 (19–20) | 20 (19–20) | 20 (16–23)b |
| D-E20 lifetime τ (ns) | 6.1 | 6.1c | 6.0 |
| D-E20-A lifetime τ (ns) | 4.7 | 5.6 | 3.9 (∼30%), 0.5 (∼70%) |
| D-E20-A FRET efficiency E | 0.23 | <0.08c | 0.33 (∼30%), 0.92 (∼70%) |
| D-E20-A D–A distance RDA (Å) | 24 (23–25) | >29 (28–30)c | 22 (18–25)b (∼30%), 13 (11–15)b (∼70%) |
- a Uncertainties due to uncertainty in κ2 (based on anisotropy) shown in parentheses. Statistical errors are shown in figures, Tables I and II.
- b Assumed value based on calculation of J on day 0–2 samples. Actual value of J between days 3 and 11 not measurable.
- c D-E20 lifetime reported as 5.6 ns in Table I although difference from monomer (6.1 ns) is not statistically significant. Table III values of E and RDA for soluble oligomers are calculated with τD = 6.1 ns are shown as an upper bound for E and lower bound for RDA.
Variation in R0 arises from changes in donor quantum yield ΦD and overlap integral J. In Table III, ΦD is shown to be equal for monomers, soluble oligomers, and aggregates at 0.26. While the value of J is nearly equal for monomers and soluble oligomers, in aggregated D-E20-A, it is not possible to calculate the true value of J. Calculation of J requires the acceptor absorbance spectrum of D-E20-A to be measured, which is not possible when significant turbidity is present. Nonetheless, the value of J for aggregated D-E20-A is considered to be similar to monomeric/oligomeric D-E20-A for the following reasons. First, the background-subtracted D-E20-A absorbance spectrum on day 2, where a small amount of aggregates are present, is identical to that on day 0, where no aggregates are present. Second, the normalized excitation ABZ donor fluorescence spectrum of D-E20 and D-E20-A remains constant over all days of the experiment (data not shown). While the excitation fluorescence spectra of the nonfluorescent YNO2 acceptor is not possible, the lack of shift of the ABZ donor absorbance spectra suggests that the YNO2 acceptor absorbance spectrum does not change as well. Third, a similar study using E20 with a tetramethylrhodamine donor at the N-terminus and Cy5 acceptor at the C-terminus revealed that the normalized fluorescence excitation spectrum of Cy5 acceptor did not shift between monomeric and aggregated samples (data not shown). While this TMR/Cy5 E20 peptide was not used due to problems with Cy5 photobleaching, it did reveal that the acceptor absorbance spectrum did not change between monomers and aggregates. Thus, it is reasonable to assume that the YNO2 acceptor absorbance spectrum does not change appreciably between monomer and aggregate states of D-E20-A.
In Table III, RET efficiencies (E) of D-E20-A in each state (monomer, oligomer, and aggregate) are converted to the D–A distance using the standard assumptions of Eqs. (7) and (8). D-E20-A monomers have a moderate end-to-end distance (24 Å). D-E20-A peptides in soluble oligomers are clearly more extended than monomers with a RET efficiency shown as E < 0.08 and RDA shown as >29 Å, with such values calculated using τD = 6.0 ns and τDA = 5.6 ns. However, the value of τD = 6.0 ns for soluble oligomers represents an upper bound of the reported value of τD = 5.6 ± 0.4 ns for SEC fractions in Table I and this bound is reflected in Table III by the presentation of RET efficiency as E < 0.08 and D–A distance as RDA > 29 Å.55 D-E20-A peptides in aggregates adopt a major population (∼70%) with a close end-to-end distance (13 Å) and a minor population (∼30%) with a moderate end-to-end distance (22 Å).
E20 Adopts Different Conformations in Monomers, Soluble Oligomers, and Aggregates
Figure 9 shows the best model of polyE/E20 aggregation, including the structural details obtained from RET in the present study. Figure 9 reveals the minimum number of states populated in the transient equilibrium achieved each day of the E20/polyE aggregation reaction. The mechanistic description in Figure 9 does not explicitly define whether soluble oligomers are kinetically on- or off-pathway from monomers and aggregates. Monomeric E20 has a moderate end-to-end distance (24 Å). E20 in soluble oligomers is more extended (>29 Å). Aggregated E20 is predominantly in collapsed conformations (13 Å) although it is possible that approximately 30% is found in conformations with a 22 Å end-to-end distance.

Model of polyE aggregation at equilibrium on day 11, consisting of 10% partially α-helical monomers (purple, 24 Å), 20% soluble oligomers (green, >30 Å) and 70% large aggregates (blue, 13 and 22 Å).
The collapsed conformation of aggregated E20 is a somewhat unexpected result as many high-resolution structures of smaller aggregated peptides (<9 residues) show highly extended structures in amyloid fibrils.11, 60 However, studies with larger peptides Aβ1–40 and Aβ1–42 show the two extended β-sheet regions within the aggregated peptide can fold back on themselves at turn regions making residues distant in primary structure close in the tertiary fold.8, 61 Even so, the closest distance between any two residues separated by 20 residues in the Aβ1–42 PDB structure 2BEG is ∼16 Å, which is greater than the 13 Å measured for D-E20-A.61 Thus, the compactness of aggregated E20 chains can be considered to be quite significant relative to these amyloid peptides. This E20 compactness may be a characteristic of peptide conformations in amorphous aggregates, although more peptides and proteins should be studied to confirm this hypothesis.
Of equal significance, the present study offers one of the first insights into the molecular structure of peptides in soluble oligomers. Although structural details of soluble oligomers have remained elusive, one might expect the conformation of peptides in soluble oligomer states to match better with larger aggregates than monomeric precursors as (a) they may be intermediate stages in the formation of larger aggregates or (b) they are competing structures with large aggregates held together with the same forces. However, in the present study, it is interesting that the D–A distance of D-E20-A in soluble oligomers (>29 Å) is farther from aggregates (13 Å) than that of monomers (24 Å). Thus, formation of different assembly states may involve separate pathways with different competing physical interactions. Further work with RET will reveal whether highly divergent soluble oligomer structures and aggregated structures formed by the same peptide are the exception or the rule.
CONCLUSIONS
The present study used both steady-state and time-resolved RET to determine quantitative structural information, i.e., an angstrom measurement of D–A distance, of the structures of E20 within soluble oligomers and large aggregates. This study has demonstrated that steady-state RET is useful but that additionally using time-resolved RET was essential to the determination that D-E20-A aggregation proceeds initially from partially collapsed α-helical monomers (24 Å end-to-end distance) to extended (>30 Å) end-to-end conformations in soluble oligomers and mixture of moderate (13 Å end-to-end distance) and compact (13 Å end-to-end distance) D-E20-A conformations in large aggregates. Having validated this experimental approach, a number of important scientific questions can now be addressed. For example, is the aggregation pathway and structures of nonamyloid polyE different from other amyloid homopolymers such as polyglutamine?50, 62 Also, do peptide conformations associated with cytotoxic assembly states differ from more benign assembly states? Finally, the present RET study has provided quantitative end-to-end distances which can be used to test the accuracy of theoretical models and simulation parameters used to study protein aggregation. Whether to study amyloid or nonamyloid aggregation pathways relevant to disease, the combined use of steady-state and time-resolved RET has enormous potential to better understand polypeptide structures at a more detailed level.

