

Regulatory Potency of Oligomeric Proanthocyanidins (PACs) on Extrafibrillar and Intrafibrillar Collagen Mineralization
Funding: This work was supported by National Institute of Dental and Craniofacial Research (DE028194).
ABSTRACT
The therapeutic potential of plant-derived proanthocyanidins (PACs) interacting with mammalian collagen is extensive, notably in strengthening specialized extracellular matrix like the dentin matrix, crucial for reparative dental treatments. This study unveils an additional facet of PACs beyond their recognized chemical and biomechanical advantages in fibrillar collagen: specific tannins possess an inherent capability to influence collagen mineralization. By leveraging the degree of polymerization (DP) of PAC oligomers binding directly to type-I collagen, selective control over in vitro mineralization is achieved. Tetrameric PACs (DP = 4) exhibit minimal barriers to extrafibrillar mineralization, whereas dimers (DP = 2) effectively hinder nucleation/growth of surface minerals, potentially favoring intrafibrillar mineralization pathways. Additionally, our investigation highlights that bound PACs facilitate the infiltration of mineral precursors within collagen fibrils without relying on conventional process-directing agents. These findings underscore the promising utility of oligomeric PACs as a new class of plant-derived process-directing agents (PPDAs) with compelling mineral activities. While our primary focus is exploring mineralization through diverse PAC structures, establishing a translatable collagen mineralization model remains one long-term goal of this line of research.
1 Introduction
Harnessing the utility of plant biomolecules interacting with mammalian biostructures has allowed remarkable advancements in optimizing material functionality and tissue microenvironments [1]. A case in point is the biomodulation of dentin—a key component of teeth—achieved with proanthocyanidins (PACs) [2], a class of plant-derived tannins initially known for their wide pharmacological properties [3].
Dentin is a calcified tissue structured around an extracellular matrix (ECM) that includes a dense network of type-I collagen fibrils and non-collagenous components [4]. Specific PACs have demonstrated the ability to modulate the matrix's chemistry and biomechanics, resulting in a noteworthy enhancement in both viscoelasticity and resistance to collagen degrading enzymes [5-7]. This enhancement is particularly significant at the dentin–resin interface, where PACs cross-linked with collagen contribute to the quality and stability of dentin–resin bonds [8, 9], supporting contemporary dental restorations.
Although PACs provide chemical and biomechanical advantages, their potential to induce collagen mineralization remains underexplored. Such a feature could enhance PAC-modified collagen matrices by promoting self-repair via in situ remineralization. Unlike bone, dentin has limited mineral compensatory mechanisms [10], making it more vulnerable to mineral loss in conditions like caries and recurrent decay. Therefore, research has pursued strategies to remineralize mineral-depleted dentin, using biomimetic in vitro models to simulate natural biomineralization [11-13].
In both bone and dentin, collagen fibrils create the scaffold for mineral deposition, a process tightly regulated by non-collagenous proteins (NCPs) [10]. Evidence suggests that the mineralization begins outside the fibrils, with NCPs stabilizing transient mineral phases that later infiltrate the fibrils [14, 15]. Current in vitro collagen mineralization models typically use calcium phosphate (CaP) nucleation inhibitors, such as polyaspartic acid (pAsp), which mimic NCPs by stabilizing a polymer-induced liquid precursor (PILP) phase [16]. This phase infiltrates collagen, transforming into oriented apatite crystals [17].
Although PILPs generated in supersaturated solutions are effective for intrafibrillar mineralization, they do not act in the extrafibrillar counterpart, thereby hindering the deposition of apatite nanocrystals between collagen fibrils [18]. More comprehensive models must integrate both intra- and extrafibrillar mineralization to mimic the natural mineralization process seen in bone and dentin [19, 20]. To improve clinical viability, strategies that directly bind biomimetic agents to collagen show promise for enhancing mineralization [21].
The intrinsic affinity of PACs for fibrillar collagen prompts curiosity about their potential ability to induce mineralization at the biointerface. The advantages of PACs as a potential mineralization agents encompass not only their collagen cross-linking activity but also their established biomodulation effects and natural origin, both of which distinguish them from synthetic polymer additives [2, 9]. The oligomeric/polymeric nature of PACs introduces structural variations contingent on monomeric units (flavan-3ols), degree of polymerization (DP), linkage types, and interflavan bond positions [22].
This study investigates how PAC oligomers, specifically dimers (DP = 2), trimers (DP = 3), and tetramers (DP = 4), influence the collagen mineralization in vitro. We assessed mineral induction in both metastable simulated body fluid (SBF) and supersaturated CaP medium, focusing on PAC structural diversity rather than developing a definitive model of collagen mineralization.
2 Materials and Methods
2.1 Materials
The primary reagent used to synthesize collagen fibrils was a commercial solution of soluble atelocollagen at 3 mg/mL, PureCol type-I collagen solution (≥ 99.9%, Advanced BioMatrix, catalog 5005). For experiments requiring a conventional nucleation inhibitor, we utilized the acidic polymer poly-L-aspartic acid sodium salt (Alamanda Polymers, catalog 000-D200) with an average molecular weight of 27 kDa. All salts for preparing buffers and ionic solutions were of analytical grade and used as received without further modifications.
2.2 PAC Oligomers Sourcing, Preparation, and Purification
Crude materials obtained from three plant sources—cinnamon bark, pine bark, and cocoa seeds—were used in the scaled-up purification of unique PAC oligomers (Table 1). The procyanidins A1 (dimer A) and B1 (dimer B) were purchased from ChromaDex (catalog ASB-00016227 and ASB-00016231, respectively).
| Oligomer DP | PACBAR representation | Molar mass (g/mol) | Oligomer label* |
|---|---|---|---|
| Dimers (DP=2) |
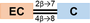
|
567.5 | A |

|
578.5 | B | |
| Trimers (DP=3) |
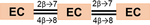
|
862.1 | AA |

|
866.2 | BB | |
| Tetramers (DP=4) |

|
1150.2 | ABA |
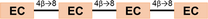
|
1154.3 | BBB | |
|
|||
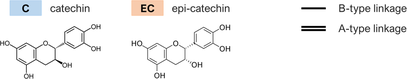
- Note: The oligomers comprised catechin and epi-catechin monomers, which are linked to form dimeric, trimeric, and tetrameric PACs. The main representative oligomers are indicated at the bottom of this table.
- * Simplified label used throughout the text considers the type (A or B) and the frequency of linkages bridging the flavanyl units.
The B-type PAC oligomers procyanidin C1 (trimer BB) and cinnamtannin A2 (tetramer BBB) were isolated from a 30-g sample of cocoa ( Theobroma cacao ) polyphenol extract (Barry Callebaut, Lebbeke-Wieze, Belgium). Briefly, the initial extract was subjected to centrifugal partition chromatography (CPC) using a solvent mixture of EtOAc/n-BuOH/MeOH/H2O (6:0.1:1:5, v/v) in ascending mode. The obtained fractions were further purified via Sephadex LH-20 in gradient elution from 70% to 100% MeOH in water, yielding trimer BB (430 mg) and tetramer BBB (300 mg). Detailed structure characterization of BB and BBB was previously documented in a publication by our research group [23].
The tetramer ABA was isolated from Pinus massoniana bark (initial extract purchased from Xi'an Chukang Biotechnology Co). Similarly, tetrameric PAC fractions were prepared from pine bark extract (200 g) by two-step CPC separation using hexane/EtOAc/MeOAc/H2O (2:4:1:4, v/v, descending) and hexane/EtOAc/MeOAc/H2O (0.4:4:1:4, v/v, ascending), further purified by reverse-phase (RP) silica gel column in gradient elution from 25% to 30% MeOH in water, yielding the tetramer ABA (900 mg). The chemical structure of ABA was determined as reported before [24].
The trimer, aesculitannin C (AA), was prepared from a previously isolated trimer, cinnamtannin B1 from Cinnamomum verum bark [25], by oxidative conversion [26]. Briefly, cinnamtannin B1 (1 g) and 2,2-diphenyl-1-picrylhydrazyl (DPPH, 1 g) were dissolved in 100 mL MeOH and stirred at room temperature. After completion of the reaction, the mixture was purified via Sephedax LH-20 column in 90% MeOH in water, resulting in trimer AA (350 mg). Detailed methods and chemical characterization of AA will be published separately. The chemical structures of the isolated compounds are given in Figure S1.
2.3 Synthesis of Type-I Collagen Fibrils
We opted for type-I collagen fibril scaffolds instead of native matrices to avoid bias from the effects of associated NCPs. The in vitro synthesis of native-like, type-I collagen fibrils followed a reliable protocol [27] with some specific adjustments. In a microfuge tube, three ingredients were combined and homogenized: 150 μL of ultrapure water, 500 μL of Na2HPO4 at 200 mmol/L with a pH adjusted to 7.3, and 250 μL of KCl at 400 mmol/L. Next, 100 μL of a commercial collagen monomer solution (3 mg/mL in 0.01 N HCl) were added to the same tube and briefly vortexed. This reaction mixture was incubated at 37°C for 4 h for spontaneous fibrillogenesis. Subsequently, the obtained type-I collagen fibril suspension was centrifuged at 9600 g for 10 min, concentrating the fibrils into a scaffold (a pellet) at the bottom of the tube. The supernatant, containing excess salts, was discarded, and ultrapure water was introduced into the tube to rinse the scaffold and remove any remaining salts. This step was repeated twice to ensure the collagen scaffold was purified and ready for further use or analysis.
2.4 PAC-Modification of Type-I Collagen Fibrils
A HEPES buffer (20 mmol/L) was employed to produce PAC oligomer solutions. Each oligomer was accurately weighed in light-protected microfuge tubes and then dissolved thoroughly in HEPES buffer to obtain oligomer solutions at 1% (w/v)—with a pH adjusted to 7 at room temperature. Oligomer solutions were used immediately to treat pristine collagen fibrils with PACs. Freshly prepared collagen scaffolds were soaked in 100 μL of the corresponding PAC oligomer solution and left for 15 min at room temperature under light protection. Next, the obtained PAC-modified collagen scaffolds were rinsed three times with ultrapure water. For the control group, collagen scaffolds were immersed in pure HEPES buffer under the same conditions as the experimental (PAC-modified) groups.
2.5 Preparation of the Mineralization Media
Three different calcium- and phosphate-containing media (A, B, and C) were prepared with the characteristics summarized in Table 2.
| Medium A | Medium B | Medium C | |
|---|---|---|---|
| Brief description | Simulated body fluid | Unstabilized CaP medium | Stabilized CaP medium |
| Initial Ca2+ concentration (mmol/L) | 2.5 | 4.5 | 4.5 |
| Initial HPO4 2− concentration (mmol/L) | 1.0 | 2.1 | 2.1 |
| Buffering agent | TBS | TBS | TBS |
| pH details | Adjusted to 7.4 at 36.5°C | Adjusted to 7.4 at 37°C | Adjusted to 7.4 at 37°C |
| Induction temperature (°C)* | 37 | 37 | 37 |
| Unbound nucleation inhibitor (100 μg/mL pAsp)? | No | No | Yes |
| Spontaneous precipitation of CaP when held at 37°C? | No | Yes | No |
- Abbreviations: pAsp, poly-L-aspartic acid; TBS, tris-buffered saline.
- * Temperature the medium was held during the mineralization study.
Medium A, also known as SBF, closely mimics the inorganic composition of human serum, excluding proteins, and does not undergo spontaneous precipitation [28]. The nonconcentrated (1×) SBF used throughout this study was prepared according to a comprehensive protocol reported by Kokubo and Takadama [29]. The as-prepared 1× SBF had its pH adjusted to 7.4 at 36.5°C and the expected ion concentrations (mmol/L) were as follows: Cl−, 147.8; Na+, 142.0; K+, 5.0; HCO3 −, 4.2; Ca2+, 2.5; Mg2+, 1.5; HPO4 −2, 1.0; and SO4 −2, 0.5.
Medium B is an unstabilized CaP solution that undergoes spontaneous precipitation and is commonly utilized in research with nucleation inhibitors [14]. Two precursor solutions were necessary to produce Medium B: a 9 mmol/L CaCl2 solution and a 4.2 mmol/L K2HPO4 solution. These precursor solutions were made using tris-buffered saline (TBS) as the solvent. The TBS solution consisted of 50 mmol/L tris-base and 150 mmol/L NaCl in water, with the pH adjusted to 7.4 at 37°C. Fresh Medium B was obtained by combining and thoroughly mixing equal parts (volume) of the calcium and phosphate precursor solutions.
For complementary experiments, a stabilized version of Medium B (namely Medium C) was produced by supplementing the calcium precursor solution with 200 μg/mL of the nucleation inhibitor pAsp (27 kDa).
2.6 Mineral Induction Procedures
The mineralization of pure and PAC-bound collagen fibrils in Medium A involved a preliminary step, “pre-soaking,” conducted as follows. Each collagen scaffold was initially soaked in 1 mL of a 150 mmol/L CaCl2 aqueous solution for 5 min. After rinsing in 1 mL of ultrapure water for 10 s, the scaffold was soaked in 1 mL of a 150 mmol/L Na2HPO4 aqueous solution for another 5 min and then rinsed again. Subsequently, the pre-soaked collagen scaffold was placed in 1.5 mL of Medium A (SBF) and incubated at 37°C. The medium was replaced by fresh SBF every 24 h to maintain a sufficient supply of mineral ions until a 96-h mineralization procedure was completed.
In the case of mineral induction in Medium B/C, collagen scaffolds were soaked solely in 10 mL of freshly prepared Medium B/C and incubated at 37°C. The mineralization was conducted for 96 h, and the medium was replaced with an equal volume every 24 h to replenish free mineral ions.
After completing the mineralization procedure in the respective medium, each collagen scaffold was thoroughly rinsed with ultrapure water, aided by low-frequency (40 kHz) sonication for 1 min in an ultrasonic cleaning bath to remove any loose mineral crystals. Rinsed samples were finally subjected to dehydration and chemical drying. For dehydration, the samples were washed in a series of graded ethanol−water solutions, including 50%, 75%, 95%, and twice in 100% (1 mL, 15 min each). Next, the samples were soaked twice in ~0.5 mL of pure hexamethyldisilazane (HMDS) for chemical drying. After the first HMDS treatment for 15 min, the supernatant was discarded, and for the second treatment, the samples were evaporated in a fume hood overnight.
2.7 Field Emission Scanning Electron Microscopy
Field emission scanning electron microscopy (FE-SEM) was performed using a Hitachi S-4800 microscope. Before analysis, the dried collagen samples were carefully transferred onto strips of conductive carbon double-sided sticky tape affixed to aluminum specimen mounts. The mounted samples were sputter-coated with a thin layer (5 nm) of Iridium. Micrographs were captured using accelerating voltages as low as 5 kV and signals from both the upper and lower secondary electron (SE) detectors. Complementary energy dispersive spectroscopy (EDS) measurements were carried out for the same samples, as detailed in the Supporting Information.
2.8 Transmission Electron Microscopy
The processing of collagen fibril samples for transmission electron microscopy (TEM) is provided in the Supporting Information. Monolayers of collagen fibrils adsorbed onto Formvar/Carbon filmed Nickel grids were analyzed without prior negative staining. The ultramorphology of these fibrils was assessed using a 300 keV transmission electron microscope, the JEOL JEM-3010, equipped with a lanthanum hexaboride (LaB6) electron source. Images were acquired with a Gatan Orius SC200 CCD camera. Fast Fourier transform (FFT) patterns were extracted from high-resolution transmission electron microscopy (HR-TEM) images.
2.9 Atomic Force Microscopy
For atomic force microscopy (AFM) imaging, pristine collagen fibrils were immobilized on freshly cleaved mica discs before undergoing the PAC modification and mineralization procedures. Details of the AFM sample preparation can be found in the Supporting Information. AFM imaging was performed in PeakForce tapping mode using a Bruker Dimension Icon microscope equipped with high-end, pre-calibrated probes (RTESPA-150-30, Bruker) with a nominal spring constant of 5 N/m. Scanning parameters were set as follows: scan rate, 0.3 Hz; scan size, 1.0 μm; and lines per image, 512. AFM measurements were taken from height images using Bruker's NanoScope Analysis 3.0.
2.10 Fourier Transform Infrared Spectroscopy
Collagen fibril scaffolds (pure collagen, PAC-bound collagen, and mineralized PAC-bound collagen) were examined for chemical composition using Fourier transform infrared (FT-IR) spectroscopy via attenuated total reflectance (ATR). The tests were conducted in triplicate on a Thermo Scientific Nicolet iS5 spectrometer equipped with a Thermo Scientific iD5 ATR accessory with a single-reflection diamond crystal. Infrared absorbance spectra were recorded in the 1400–500 cm−1 range at a resolution of 4 cm−1 using 32 accumulations.
2.11 Data Analysis
Graphs were generated using GraphPad Prism 9. Spectroscopic data normalization and band integration were performed with OriginPro 2020. Calcium-to-phosphorus (Ca/P) ratios were analyzed by one-way ANOVA, followed by Tukey's post hoc test (α = 0.05), using IBM SPSS Statistics 28.0.
3 Results and Discussion
Collagen cross-linking with plant-derived PACs is a remarkably effective chemical modification to impart enhanced biomechanics and biostability to functional collagenous matrices (Scheme 1) [2, 30].

As part of this study, we confirmed that oligomeric PACs bind to reconstituted type-I collagen under physiological pH, as reported previously for native collagenous tissues such as dentin [7]. Examining in vitro-assembled fibrils under an atomic force microscope revealed a distinct 67-nm D-periodicity (Figure 1A), characteristic of native type-I collagen [31]. However, immersion in 1% PAC solutions elicited different effects on the fibril topography, contingent upon the DP of the PAC oligomer. Larger tetramers (DP = 4) formed a homogeneous film eveloping the collagen, effectively masking its typical banding pattern (Figure 1B) and resulting in an increased fibril diameter (Figure 1C). Conversely, smaller PACs such as dimers (DP = 2) created a more subtle coating, enabling partial visualization of the underlying collagen structure (Figure S2).

Despite the morphological changes influenced by DP, the collagen–PAC complex retained its fibrillar nature, owing to a relatively lower concentration of bound PAC molecules compared to the proteinaceous matrix. This inference aligns with the FT-IR results, where essential IR frequencies specific to type-I collagen, such as the prominent amide I peak at ~1650 cm−1, remained predominant after treatment (Figures 1D and S3). Nevertheless, the cross-linking of flavanyl moieties is discernible through the broadening of the amide II band at ~1550 cm−1 (Figure 1D), indicative of hydrogen bonding between the PACs and the free amino groups (NH2) of collagen [32]. Moreover, noticeable shifts in multiple IR bands toward slightly lower frequencies following PAC treatment strongly suggest biomodification effects [7].
The ability of PAC-modified collagen to induce mineral formation in vitro was assessed using different CaP solutions (Table 2), and the findings regarding structure–activity relationships (SARs) to biomimetic mineralization are discussed as follows.
3.1 Contribution of Bound PACs to Unconfined Nucleation in Type-I Collagen Fibrils
In assessing the ability of modified collagen to induce mineral growth “extrafibrillarly”, it was crucial to consider a CaP solution that does not support homogeneous nucleation, that is, the formation of nuclei from clear liquids. Non-concentrated (1×) SBF was an ideal choice, since this biomimetic solution can sustain its metastable state even at 37°C [29].
To achieve dominant extrafibrillar mineralization in 1× SBF, we implemented a protocol detailed in our previous publication [33]. This involved pre-soaking the matrix in concentrated calcium and phosphate solutions before its incubation in SBF. Pre-soaking, recognized as a mineral induction technique [34], enhances local supersaturation and is especially advantageous for substrates like type-I collagen, which lack the intrinsic ability to nucleate apatite in non-concentrated SBFs.
Collagen modified with PACs was compared against densely mineralized untreated collagen (Figure 2A–C) as a reference for extrafibrillar mineralization. A high-magnification view of the mineralized fibrils reveals aggregates of CaP nanosheets (Figure 2C), in agreement with the morphology of apatite crystals expected to form when using the SBF immersion technique [35].

The investigated PACs did influence mineral induction of (pre-soaked) PAC-bound collagen in 1× SBF. The contribution of their DP to the unconfined (extrafibrillar) nucleation was evident, where DP affected the morphology and distribution of CaP polymorphs within the fibril network (Figure 2D–F). Smaller oligomers such as dimer B inhibited the surface-directed mineralization process, and the fibril web was virtually devoid of extrafibrillar minerals (Figure 2D). The opaque nature and “corn-on-the-cob” texture of the fibrils (Figure S4) suggest intrafibrillar mineralization, as previously noticed [36]. In the vicinities of collagen fibrils cross-linked with the trimer BB, spherical nanoparticles formed as mineral precursors (Figure 2E). Finally, when using larger oligomers such as tetramer BBB, plentiful plate-like crystals aggregated along the fibrils (Figure 2F), recapitulating the morphology and distribution of the control group's apatitic coating.
The SARs concerning the DP of PACs were identified not only for the aforementioned B-type PACs but also for analogs of the A-class, including dimer A, trimer AA, and tetramer ABA (Figure S5). Note that the type of interflavanyl linkages, that is, CC bond (B-type) or CC plus ether bonds (A-type), did not influence the mineral induction in SBF.
EDS data demonstrate that, after the immersion in SBF, all PAC-bound collagen scaffolds exhibited the presumed calcium and phosphorus elements (Figure S6). However, the elemental composition of the extrafibrillar mineral varied (from the slurry attached to dimer-bound fibrils to apatitic crystals around tetramer-bound fibrils), and the Ca/P molar ratio tended to increase with the DP (Figure 3). Spherical nanoparticles associated with trimer-bound collagen presented low (< 1) Ca/P ratios, suggesting mixed intermediate phases [37]. On the other hand, fully developed polymorphs around tetramer-bound fibrils showed Ca/P ratios between 1.5 and 1.6, which fall within the range of biological apatites [38].

From the perspective of another chemical property of tannins, oligomeric PACs are likely to exert an influence on extrafibrillar mineralization due to their innate capacity to chelate metal ions. The ability of PACs to coordinate with various transition metals, such as Cu, Fe, and Zn, is well-documented, and this coordination is harnessed across diverse industrial and biological applications [39, 40]. It is, thus, plausible that the generation of calcium–polyphenol complexes may govern the CaP nuclei formation dynamics on a PAC-bound surface. However, it is important to note that the ability of PAC oligomers to sequester calcium ions does not necessarily facilitate CaP nucleation. Polyanionic polymers, such as pAsp, also sequester mineral ions but inhibit rather than promote CaP nucleation [14]. This provides a rationale for the observed gradual reduction of unconfined nucleation in SBF as the DP decreased.
Although pre-soaked tetramer-bound collagen offered minimal resistance to unconfined nucleation in SBF, the dimers restrained this process. This apparent discrepancy is likely due to the molecular size of PAC oligomers (proportional to DP) and the associated count of accessible chelating sites. Nevertheless, indirect variables cannot be dismissed, including the DP-dependent viscoelastic behavior of the modified substrate. For instance, Reis and colleagues [7] have shown that dentin matrices cross-linked using tetrameric PACs are stiffer (present higher storage modulus) than those cross-linked with dimers. This tunability in the mechanical attributes of the collagenous matrix can potentially affect its aptitude for apatite formation [41].
Interestingly, it was possible to counteract specific inhibitory effects of PACs on unconfined nucleation by boosting the initial mineral induction via pre-soaking. Trimer-bound collagen fibrils incubated in 1× SBF after three pre-soaking cycles showed a rich mineral coverage of plate-like crystals—as opposed to nanoprecursors dotting the surface of fibrils pre-soaked only once (Figure 4).

The results in Figure 4 suggest that the metal ion–PAC interaction and subsequent nucleation is a progressive process influenced by mineral ion saturation. It appears that calcium ions coordinated with (bound) PACs may not readily react with free HPO4 2− to initiate the formation of a new bulk phase. In this case, the effectiveness of pre-soaking—as a strategy to increase local saturation of mineral precursors [42]—is attenuated, thereby hindering further apatite growth in nonconcentrated SBF.
3.2 Contribution of Bound PACs to Confined Nucleation With and Without Unbound Nucleation Inhibitors
The varying inhibitory strengths of bound PACs toward unconfined nucleation prompted an essential query: By restricting crystal growth on the collagen surface, can PAC oligomers promote conditions conducive to confined (intrafibrillar) nucleation? To examine this hypothesis, we evaluated the induction behavior of PAC-bound collagen in Medium B (Table 2). This medium, with calcium and phosphate levels approximately double those in human serum, typically induces spontaneous precipitation in bulk solution at physiological temperatures, unless counteracted by a nucleation inhibitor such as pAsp [14]. Its slightly higher supersaturation toward apatite proves advantageous for achieving nucleation upon confinement of precursor phases. Notably, analogous medium has been effectively employed in investigating the activity of macromolecules bound to collagenous matrices [21].
In the unstabilized medium, CaP precipitated randomly as particle aggregates in the vicinities of untreated (non-modified) collagen fibrils (hollow pointer in Figure 5D), as expected [18]. On the other hand, PAC-modified fibrils presented a pronounced fibrillar texture and reduced amounts of loose extrafibrillar deposits (Figure 5A–C). Bare regions of untreated collagen (white pointer in Figure 5D) looked semi-transparent under TEM (white pointer in Figure 5H) because of the lack of a highly electron-dense inorganic phase in the internal compartments. Conversely, PAC-modified collagen fibrils revealed contrasting dark regions that marked the banding pattern (pointers in Figure 5E–G), corroborating the confined nucleation.

Collagen banding discernible under unstained TEM is a distinctive sign of intrafibrillar minerals, where the tropocollagen density echoes with the hierarchical arrangement of electron-dense apatite crystallites [13, 43]. As the banding pattern was discrete in the representative samples (Figure 5E–G), we concluded that the intrafibrillar inorganic phase did not attain complete crystallization during the 96-h incubation in Medium B. A mineral phase at least partially crystalline was verified for fibrils modified with dimer B (Figure 6). As deduced from FFT patterns, the other B-type oligomers induced an amorphous CaP phase within the collagen's interstices (Figure S7).

The partial intrafibrillar mineralization evident in PAC-bound fibrils may be attributed to several factors, including limited availability of precursor ions/clusters within Medium B. Without an unbound nucleation inhibitor like pAsp, spontaneous precipitation occurs over time, shifting the equilibrium toward insoluble CaP. Consequently, dissociated species become less available to permeate the interstices of the fibril or form prenucleation clusters at the PAC-bound surface. This scarcity of precursor ions in unstabilized media possibly explains why certain biomimetic analogs, for example, sodium trimetaphosphate [43], fail to promote intrafibrillar mineralization when bound to collagen rather than being supplemented in the medium.
Addtitionaly, it has been suggested that macromolecules smaller than 6 kDa can traverse the collagen fibril structure [44]. Given the notably smaller size of PAC oligomers (Table 1) in comparison to bulky polymer additives like pAsp (as bulky as 27 kDa), it is conceivable that these oligomers infiltrate the compartments of collagen during the biomodification process. Should this hold true, the oligomers could exert their chelating effect “intrafibrillarly,” affecting the progression of crystallization in the confined spaces.
In addition to the B-type PACs addressed above, evidence of intrafibrillar mineral induction was verified for collagen modified with an A-type analog (Figure S8). Reinforcing the formation of intrafibrillar minerals, the Young's moduli of individual PAC-bound collagen fibrils exhibited an increase corresponding to longer incubation times in Medium B (Figure S9).
In contrast to the sampling of randomly crushed fibril fragments for TEM, whole collagen scaffolds were scanned using FT-IR spectroscopy. The compositional analysis showed that PAC-modified collagen scaffolds were calcified after extended incubation in Medium B (Figure 7). Only bands typical of the organic constituents were noted in the absorbance spectra of samples scanned before incubation (dotted lines in Figure 7). Typical instances are the prominent peaks of amide I (CO stretch) at ~1650 cm−1 and amide II at ~1550 cm−1, as well as the moderate absorptions from amide III in the 1300–1180 cm−1 range [45]. After incubation, bands commonly observed in apatitic minerals appeared in the spectra for PAC-modified collagen (solid lines in Figure 7B–D). The wide band at ∼1022 cm−1 was assigned to the ν3 PO4 asymmetric stretching, while the ν1 PO4 appeared as a discrete shoulder at ∼960 cm−1. A thin band at ∼875 cm−1 was due to the POH stretching, detectable in non-carbonated apatites [46]. Since Medium B had a simplified formulation devoid of carbonate salts, common carbonate substitutions in the mineral component were not observed.

The mineral-to-matrix ratios were determined by calculating the band area ratio of ν3 PO4 to amide I. Following an extended incubation period in Medium B, it was noted that the mineral-to-matrix ratio was elevated for PAC-modified samples (COL–B, 2.49 ± 0.33; COL–BB, 1.26 ± 0.39; COL–BBB, 0.67 ± 0.03) in comparison to untreated collagen (COL, 0.24 ± 0.04). The observed higher ratios reinforce that more inorganic phase was incorporated into the interstices of PAC-modified fibrils.
Complementary tests indicated that the PACs were compatible with the pAsp-mediated intrafibrillar mineralization approach. Using Medium C, which is a variation of Medium B containing pAsp at a typical concentration (100 μg/mL), confined nucleation was dominant in both untreated collagen fibrils and collagen modified with PACs (Figure S10).
Overall, the present findings suggest that the selected PACs are promising process-directing agents that can lead to phase selection at the collagen–PAC interface. By modifying collagen chemistry and biomechanics, the structurally diverse (DP, linkage types) PAC oligomers favored nucleation within the confined spaces using a CaP medium devoid of crucial polymer additives.
Another broader conclusion is that PACs are unlikely to strictly inhibit CaP nucleation in solution. Classic inhibitors such as pAsp sequester calcium and phosphate ions to form highly hydrated complexes that behave like a liquid, known as the PILP phase [17]. Unlike PILPs, some tested PACs interacted with free mineral ions, generating solid complexes. When Medium B was supplemented with PAC oligomers (without collagen), spontaneous precipitation of particles still occurred over time (Figure S11). However, the recovered CaP–PAC complexes were remarkably smaller than apatite crystals generated in a PAC-free medium, attesting to the PAC activity.
Based on the new data, we hypothesize that the modulation of PACs on intrafibrillar mineralization starts at the surface level, where the metal chelation ability of immobilized oligomers leads to the selection of transient, amorphous mineral precursors, while preventing surface-directed nucleation. These precursors can then seep into the water-filled compartments within the collagen fibril. Nucleation and crystallization likely depend on factors such as medium saturation and inclusion/exclusion of PAC oligomers in the collagen's compartments. This potential mechanism aligns with one of the prevailing mineralization theories, which suggests that mineralization is facilitated through the formation of prenucleation clusters that infiltrate the collagen's interstices via electrostatic interactions [47].
Further investigations could elucidate how bound PACs affect the propagation of the mineral phase through the collagen fibrils and its transformation over time. The present study, conducted with isolated collagen fibrils, also revealed certain limitations that open opportunities for future exploration: one such area lies in elucidating the nature of the mineral phase with extended incubation times.
4 Conclusions
Bound PAC oligomers afford selective control over extrafibrillar mineralization within type-I collagen through an SBF-immersion technique. The DP emerges as a pivotal factor, determining the level of inhibition in unconfined nucleation. Specifically, tetrameric PACs (DP = 4) exhibit minimal barriers to unconfined nucleation, whereas dimers (DP = 2) effectively hinder nucleation/growth of surface minerals. This hindrance potentially facilitates intrafibrillar mineralization pathways.
Under saturation conditions higher than in SBF, the bioactivity of bound PACs becomes essential for mineral induction in the confined compartments of collagen. Notably, the PAC-mediated confinement of the mineral phase occurs independently of typical polymer additives like pAsp in the unstabilized medium. However, the prevailing amorphous nature of intrafibrillar minerals under specific experimental conditions warrants further exploration. Resolving the limited crystallization of intrafibrillar minerals is pivotal for drawing conclusive insights into regulating intrafibrillar versus extrafibrillar dynamics.
Our findings underscore the promising utility of oligomeric PACs as a new class of plant-derived process-directing agents (PPDAs) with compelling mineral activities. This work sheds some new light on the mineral induction ability of a particular class of phytochemicals that hold promise for the stabilization of collagen in vulnerable ECMs (e.g., apatite-depleted dentin). While our primary focus was exploring mineralization through diverse PAC structures, establishing a translatable collagen mineralization model remains an ultimate goal of this line of research.
Author Contributions
Odair Bim-Junior: conceptualization (equal), data curation (lead), formal analysis (lead), writing – original draft (lead). Shuxi Jing: data curation (supporting), formal analysis (supporting), investigation (supporting), writing – review and editing (supporting). James B. McAlpine: formal analysis (supporting), methodology (supporting), writing – review and editing (supporting). Shao-Nong Chen: data curation (supporting), formal analysis (supporting), funding acquisition (supporting), project administration (supporting), writing – review and editing (supporting). Guido F. Pauli: conceptualization (supporting), formal analysis (supporting), funding acquisition (supporting), project administration (equal), writing – review and editing (equal). Ana K. Bedran-Russo: conceptualization (lead), formal analysis (equal), funding acquisition (lead), project administration (lead), writing – review and editing (lead).
Acknowledgments
This study was supported by the National Institute of Health (NIH, grant # R01DE028194). We thank Dr. Heather Owen and Milad Momtaz for guidance with electron microscopy and imaging assistance.
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

