

Discrimination of the Enantiotopic Faces of Structurally Unbiased Carbenium Ions Employing a Cyclohexadiene-Based Chiral Hydride Source
Graphical Abstract
A chiral cyclohexadiene-based dihydrogen surrogate can distinguish between the enantiotopic faces of benzylic carbenium ions (see Scheme). No covalent interactions are required but instead dispersion controls the facial discrimination as verified by computations.

Abstract
An enantioselective reduction of simple carbenium ions with cyclohexadienes containing a hydridic C−H bond at an asymmetrically substituted carbon atom is disclosed. The net reaction is a transfer hydrogenation of alkenes (styrenes) only employing chiral cyclohexadienes as dihydrogen surrogates. The trityl cation is used to initiate a Brønsted acid-promoted process, in which a delicate intermolecular capture of a carbenium-ion intermediate by the aforementioned chiral hydride source is enantioselectivity determining. Exclusively non-covalent interactions are rendering one of the transition states energetically more favored, giving the reduction products in good enantiomeric ratios. The computed reaction mechanism supports the present findings as well as previous results obtained from studies on other transfer-hydrogenation methods involving the cyclohexadiene platform.
Carbocations are fundamentally important reactive intermediates in synthetic chemistry1 and key to the mechanistic understanding of a broad spectrum of cationic transformations.2-5 Their different forms are typically classified into tricoordinate carbenium ions and non-classical, pentacoordinate carbonium ions. Two general approaches for their synthesis exist (Scheme 1, top): by abstraction of a leaving group (left) or by protonation of a C−C double bond (right). In the given examples, a benzylic and as such stabilized carbocation is generated.3 As a result of their fleeting nature even at low temperature, discrimination between the enantiotopic faces of prochiral, structurally unbiased carbocations by intermolecular capture with a nucleophile remains a daunting challenge. However, a few successful strategies have emerged in the past decades (Scheme 1, bottom).6 Bach introduced diastereoselective inter- and intramolecular reactions of carbocations having an adjacent stereogenic center with aryl nucleophiles [Eq. (1)].7 In a subsequent report, Chung observed an inverted selectivity by employing phenyl substituents at the stereogenic center.8 Recently, the directing effect of chiral counteranions such as phosphoric acid derivatives9 was exploited for the enantioselective nucleophilic attack at benzylic or propargylic carbenium ions [Eq. (2)]. Of note, List reported the first enantioselective cyclization initiated by protonation of prochiral alkenes.10 Jacobsen11 and Carreira12 disclosed approaches with a chiral hydrogen-bond donor to coordinate the counteranion and a chiral metal complex to induce enantiofacial control, respectively (not shown). Also, non-classical carbocations have been employed in asymmetric synthesis, and List introduced a strategy that enabled enantioselective Friedel–Crafts reactions of the norbornyl cation in good yields and with high enantiomeric ratios (not shown).13

Methods for the generation of prochiral carbenium ions and trapping with nucleophiles. Ar=aryl, R=aryl or alkyl, LG=leaving group, Nu=nucleophile, RS=small group, RL=large group, [A*]−=chiral anion, αNp=naphth-1-yl.
Prior to that work, Fry employed silicon-stereogenic hydrosilanes as hydride sources to achieve enantiofacial differentiation [Eq. (3)].14 Although only little enantiomeric excess was seen in this series of seminal experiments, it is an early (if not the first) example of achieving enantioinduction in a reagent-controlled trapping of a prochiral carbenium ion.15, 16 Intrigued by these advances, we set out to design a hydride source that enables the discrimination of the enantiotopic faces of carbenium ions in an intermolecular, reagent-controlled reaction. For this, we turned our attention towards B(C6F5)3- and Brønsted acid-catalyzed transfer-hydrogenation reactions of unactivated alkenes employing cyclohexadienes as dihydrogen surrogates previously introduced by our laboratory.17, 18
These reactions are outlined in Scheme 2 (top) and are characterized by two elemental steps: protonation of alkene I followed by hydride transfer onto the intermediate carbenium ion II. When directly using a Brønsted acid for alkene protonation, the cyclohexadiene III acts as the hydride source (II→V, right cycle). The Wheland complex IV (gray circle) thus generated subsequently maintains the catalytic cycle either acting as the Brønsted acid or regenerating the acid HA (IV→VI). The situation is more complex with the boron Lewis acid because B(C6F5)3 could be the catalyst or just an initiatior (left cycle).19 B(C6F5)3 engages in hydride abstraction from the cyclohexadiene III to form the aforementioned Wheland intermediate IV along with the corresponding borohydride. That borohydride could in principle either compete with the cyclohexadiene III in carbenium-ion reduction17b or simply be a non-participating counteranion. This ambiguity disqualifies the B(C6F5)3-catalyzed transfer hydrogenation as a starting point for the development of a reagent-controlled variant with a chiral cyclohexadiene reagent. Hence, initiation by a Brønsted acid or a trityl salt such as [Ph3C][B(C6F5)4] is required (Scheme 2, bottom).

Lewis and Brønsted acid-catalyzed transfer hydrogenation of alkenes. R1 and R2=aryl or alkyl with one typically being an aryl group.
We began our investigation by identifying a synthetic route to enantioenriched dihydrogen surrogates.20 Although asymmetric versions of the Birch reduction are known, the substitution patterns would not allow for the synthesis of dihydrogen surrogates.21 An alternative strategy to the targeted chiral cyclohexadienes is an asymmetric cycloaddition, constructing the cyclohexadiene core with the desired hydridic C−H bond at an asymmetrically substituted carbon atom in one synthetic operation. Perfectly suited rhodium-catalyzed formal [4+2] cycloadditions were developed recently. First introduced by Hayashi22 and further optimized by Shibata,23 Zheng24 disclosed a procedure with good overall yield and excellent enantioselectivities in selected cases.25 Employing a cationic rhodium catalyst in combination with the chiral phosphoramidite ligand L*, the desired cyclohexadienes became accessible (Scheme 3). With this method in hand, various substituted dihydrogen surrogates 3 aa–ca/ac were synthesized from alkyl-substituted dienes 1 a–c and acetylene dicarboxylates 2 a–c in moderate to good yields and with good to high levels of enantioselection. An exclusively alkyl-substituted cyclohexadiene 3 ad was prepared in two steps from dihydrogen surrogate 3 aa (see the Supporting Information for details). The absolute configuration of those surrogates was assigned as R by comparison of the direction of the optical rotation of 3 aa with a reported value.25

Synthesis of chiral dihydrogen surrogates 3. nbd=norbornadiene.
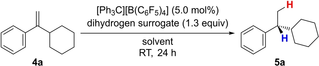
We next turned to the asymmetric transfer hydrogenation of the unactivated model alkene 4 a and decided on the use of the trityl salt [Ph3C][B(C6F5)4] as an initiator for the reaction (Table 1). A longer reaction time (48 h) and incomplete conversion was observed when using the Brønsted acid Tf2NH; of note, B(C6F5)3 does not promote the reaction. After abstraction of a hydride from the dihydrogen surrogate, the resulting Wheland intermediate then acts as a Brønsted acid, entering the Brønsted acid-catalyzed cycle outlined in Scheme 2 (right).17b Dihydrogen surrogate 3 aa afforded the product 5 a in moderate yield and enantioselectivity (entry 1). A methyl substituent in the bisallylic position gave lower and a nonyl substituent slightly better yield and enantioselectivity (entries 2 and 3). Sterically more demanding esters as in 3ab led to a longer reaction time (entry 4). The n-hexyl-substituted derivative 3 ac gave the best selectivity while maintaining a sufficiently high yield (entry 5). Dihydrogen surrogate 3 ad devoid of the carboxyl groups brought about an erosion of the enantioselectivity (entry 6). This finding led to the hypothesis that the proximal ester group plays a crucial role in the preorganization of the carbenium ion prior to the hydride transfer. Replacing CH2Cl2 by halogenated aromatic solvents (entries 7 and 8; for FC6H5 and 1,2-F2C6H4, see Table S2 in the Supporting Information) or benzene and toluene gave higher enantiomeric ratios (entries 9 and 10). Toluene showed the best results; no reaction was observed at 0 °C. The reaction did neither proceed in a hydrocarbon nor an ethereal solvent (entries 11 and 12). In turn, moderate conversion but low enantioselection was found in polar and polar protic solvents such as nitromethane and HFIP, respectively (entries 13 and 14).
|
||||
Entry |
Dihydrogen surrogate |
Solvent |
Yield [%][c] |
e.r.[d] |
|---|---|---|---|---|
1 |
3 aa |
CH2Cl2 |
67 |
78 : 22 |
2 |
3 ba |
CH2Cl2 |
30 |
67.5 : 32.5 |
3 |
3 ca |
CH2Cl2 |
73 |
80 : 20 |
4[e] |
3 ab |
CH2Cl2 |
91 |
81.5 : 18.5 |
5 |
3 ac |
CH2Cl2 |
84 |
82 : 18 |
6 |
3 ad |
CH2Cl2 |
84 |
54 : 46 |
7 |
3 ac |
ClC6H5 |
75 |
86.5 : 13.5 |
8[f] |
3 ac |
1,2-Cl2C6H4 |
85 |
84 : 16 |
9 |
3 ac |
benzene |
88 |
88 : 12 |
10 |
3 ac |
toluene |
89 (78)[g] |
88 : 12 |
11 |
3 ac |
n-pentane |
– |
– |
12 |
3 ac |
Et2O |
– |
– |
13 |
3 ac |
MeNO2 |
56 |
73 : 27 |
14 |
3 ac |
HFIP |
31 |
60 : 40 |
- [a] All reactions were performed on a 0.1 mmol scale in the indicated solvent. [b] See the Supporting Information for the complete optimization. [c] Yield determined by GLC analysis with n-decane as an internal standard. [d] Determined by GLC analysis on a chiral stationary phase. The absolute configuration was assigned as R by comparison with reported data.26 [e] 48 h reaction time. [f] An almost identical reaction outcome (75 % yield and an e.r. of 84 : 16) was obtained with [Ph3C][HCB11H5Br6] as the initiator. [g] Isolated yield in parentheses. HFIP=1,1,1,3,3,3-hexafluoroisopropanol.
We then set out to explore the substrate scope (Scheme 4, top). Although initially optimized for a simple phenyl-substituted alkene, we found that 4-methoxy substituted alkenes 4 b–j afford the products in higher yields and with better enantiomeric ratios. This observation can be rationalized by the better stabilization of the intermediate carbenium ion leading to a prolonged lifetime and potentially improved preorganization of the reactants. Of note, a para-dimethylamino group was not compatible as were an ortho-tolyl or a mesityl group (not shown). The alkene 4 b bearing a cyclohexyl group smoothly converted into alkane 5 b with higher yield and e.r. compared to 5 a. An effect of the ring size was observed as a five- and seven-membered cycloalkyl groups led to diminished yields and enantioselection (5 c and 5 d). Neopentyl, methyladamantyl and benzyl groups were all tolerated, and the alkenes 4 e–g yielded the corresponding products 5 e–g. An ester in the sidechain of alkene 4 h was accepted but a modest enantiomeric ratio was obtained. The geminal allyl dichloride 4 i was tolerated, and it was only the indene derivative 4 j that showed no enantioinduction after its hydrogenation. Sterically demanding substrates such as 4 k showed no reactivity towards this transfer hydrogenation (Scheme 4, bottom). Electron-deficient alkene 4 l failed to react, corroborating our hypothesis that the stability of the carbenium ion is essential for this transfer process. Double bond isomerization as a side reaction under the Brønsted acidic reaction conditions was seen in a few cases; alkene 4 m gave the isomerized, conjugated starting material without further reduction of the internal alkene. For most other substrates, isomerization prior to the transfer hydrogenation followed by protonation does not interfere since this reaction sequence arrives at the same carbenium-ion intermediate as the direct protonation of the initially used alkene.

Substrate scope I of the asymmetric transfer hydrogenation. All reactions were performed on a 0.1 mmol scale. Enantiomeric ratios were determined by GLC or HPLC analysis on chiral stationary phases. [a] 60 % yield and an e.r. of 89.5 : 10.5 were obtained on a 1.0 mmol scale; 61 % yield and an e.r. of 90 : 10 were obtained in 1,2-Cl2C6H4 with [Ph3C][HCB11H5Br6] as the initiator. [b] The absolute configuration of these products was assigned with reported literature data (see Supporting Information for details), and that of the other products in analogy. Ad=adamant-1-yl.
This assumption was verified by subjecting the terminal and internal alkenes 4 n and 4 n′ to the reaction conditions (Scheme 5, top). In both cases product 5 n did indeed form in similar yields and enantiomeric ratios. Racemic substrates 4 o and 4 p bearing a stereogenic carbon atom in the allylic position (not shown) furnished the corresponding products 5 o and 5 p, respectively. This resembles the Bach-type systems,7 yet the diastereocontrol of approximately 2 : 1 is less pronounced for the hydride transfer. The high isolated yields indicate that preceding alkene isomerization is occurring, and the stereochemical outcome is further blurred by kinetic resolution effects. The allylic bromide 4 q failed to react and did not afford product 5 q (Scheme 5, bottom). Conversely, the corresponding vinylic bromide 4 q′ was transformed into 5 q in moderate yield and enantiomeric ratio. This stands in contrast to existing hydrogenation methods since transition-metal-catalyzed procedures are often burdened with hydrodehalogenation as a side reaction.27

Substrate scope II of the asymmetric transfer hydrogenation. All reactions were performed on a 0.1 mmol scale. Enantiomeric ratios were determined by GLC or HPLC analysis on chiral stationary phases. Diastereomeric ratios were determined by GLC analysis. [a] The relative configuration was assigned based on reported literature data (see Supporting Information for details).
To gain mechanistic insight into the experimentally observed enantioinduction in this reagent-controlled reduction of unbiased carbenium ions, state-of-the-art dispersion-corrected DFT calculations were performed at the PW6B95-D3/def2-QZVP+COSMO-RS level of theory using TPSS-D3/def2-TZVP+COSMO optimized geometries.28 The trityl cation [Ph3C]+ was used as the initiator, and cyclohexadiene 3 da and alkene 4 a were chosen as reactants in toluene solution as a representative chemical system. The final free energies in kcal mol−1 at 298 K in 1 M concentration in toluene are used in the discussion.
As shown in Figure 1, the reaction is initiated by hydride transfer from cyclohexadiene 3 da to cation [Ph3C]+, which is 4.8 kcal mol−1 endergonic over a sizeable barrier of 24.2 kcal mol−1 (via transition state TS1+). The resulting arenium ion 6 da+ is the reactive proton source. No stable adduct of [Ph3C]+ at either ester carbonyl oxygen atom of 3 da could be found. The alkene substrate 4 a is then protonated by 6 da+ at the terminal carbon atom over a very low barrier (via transition state TS2+).29 This step is highly exergonic, reaching the carbenium ion 8 a+ that is more reactive than the initiator, i.e. the trityl cation [Ph3C]+, with respect to further hydride abstraction from the chiral cyclohexadiene reagent 3 da. Indeed, (R)-selective hydride transfer from 3 da to 8 a+ (via transition state TS3R+) is now −5.0 kcal mol−1 exergonic over a low barrier of only 8.8 kcal mol−1. Mainly due to higher steric hindrance, the competing (S)-selective hydride transfer (via transition state TS3S+) is kinetically less favorable by 1.1 kcal mol−1 with a significant contribution of 0.3 kcal mol−1 from London dispersion interactions. This is in good agreement with the experimentally obtained enantiomeric ratio of product 5 a; an energy difference of 1.1 kcal mol−1 corresponds to e.r.=86 : 14. A closer examination of this hydride transfer process shows that the chiral cyclohexadiene-based surrogate 3 da and the prochiral carbenium-ion intermediate 8 a+ are brought together by favorable dispersion interactions of about 18 kcal mol−1 mainly between the cyclohexadiene ring and the phenyl ring to arrive at those transition structures. These are, however, disfavored by solvation effects by about 11 kcal mol−1. These data indicate the crucial role of dispersion interactions in enabling such hydride transfer reactions even in solution.

Gibbs free energy profile of the reagent-controlled alkene transfer hydrogenation. In kcal mol−1 at 298 K and 1 M in toluene solution, computed at the PW6B95-D3/def2-QZVP+COSMO-RS level of theory using TPSS-D3/def2-TZVP+COSMO optimized geometries. Initiated by hydride transfer from cyclohexadiene 3 da to cation [Ph3C]+ (blue line), alkene substrate 4 a is protonated by the arenium ion 6 da+ to form the carbenium ion 8 a+, followed (R)-selective hydride abstraction from 3 da to reach the (R)-configured product 5 a and regenerated Wheland intermediate 6 da+.
Moreover, we also inspected our initial assumption that the proximal ester group is the salient feature of these chiral hydride sources by DFT calculations (cf. 3 ad with two methyl groups; Table 1, entry 6). Indeed, we identified a strong electronic interaction of 21.9 kcal mol−1 between the carbocation 8 a+ and the ester carbonyl oxygen atom of 3 da (see Table S3 in the Supporting Information) that is however disfavored by solvation and entropy effects by 11.2 and 18.0 kcal mol−1, respectively. Hence, this carboxonium-ion formation becomes 5.4 kcal mol−1 unstable in solution but is as such 3.4 kcal mol−1 lower in free energy than the hydride transfer transition state TS3R+. On the basis of this insight, we conclude that the ester group itself is not essential but, while that Lewis adduct is unlikely a productive intermediate, it helps preorganizing the reactants 8 a+ and 3 da before the hydride transfer via TS3R+. Other bulky substituents could fulfill the same role as the carboxyl group.
In summary, we introduced here an asymmetric hydride transfer process based on the use of the cyclohexadiene platform. In the key step of this reaction, the enantiotopic faces of structurally unbiased carbenium ions are discriminated by trapping with that chiral hydride source. Computations indicate that the transition states of that enantioselectivity-determining step differ in their free energies by 1.1 kcal mol−1 due to varying non-covalent interactions, mainly consisting of π-π-stacking and dispersion. The products of this reagent-controlled transfer hydrogenation are isolated in acceptable yields and, given the origin of the enantioinduction, with good enantioselection.
Acknowledgments
This research was (in part) supported by the Deutsche Forschungsgemeinschaft (Oe 249/18-1) as well as TU Berlin. M.O. is indebted to the Einstein Foundation Berlin for an endowed professorship. Z.-W.Q. and S.G. are grateful to the Deutsche Forschungsgemeinschaft (project 490737079) for financial support. We thank Valentin Poirier, Sophie C. Schulze, and Paul E. Rucker for their experimental contributions as well as Dr. Tao He for the donation of [Ph3C][HCB11H5Br6] (all TU Berlin). Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

