

Facile Synthesis of Nitrogen-Doped Nanographenes with Joined Nonhexagons via a Ring Expansion Strategy
Graphical Abstract
In this study, novel nanographenes bearing N-doped pentagon-heptagon pairs were synthesized through a facile ring expansion strategy. Modifications of heptagon spacers have endowed N-doped nanographenes with tunable physicochemical properties. This ring expansion method was further explored to synthesize diverse N-doped nanographenes containing pentagon-octagon pairs, demonstrating the high efficiency of the proposed method.

Abstract
Synthetic methodology is considered a holy grail in both organic chemistry and materials science. Over the past few decades, researchers have explored graphene-type molecules (or nanographenes) through classic Scholl oxidative cyclodehydrogenation. Despite the successes achieved with various nanographenes, the development of new methods to synthesize these highly desired molecules lags behind. Herein, we developed a facile and effective method to produce a series of nanographenes bearing nitrogen (N)-doped pentagon-heptagon pairs in acceptable yields. Modification of the heptagonal ring endowed the resultant nanographenes with tunable physicochemical properties; for instance, M9 exhibited both aggregation-caused quenching and aggregation-induced emission behavior. Most strikingly, novel nanographenes containing N-doped pentagon-octagon pairs were also obtained using the same synthetic strategy, demonstrating the superior versatility and efficiency of the proposed ring expansion method.
Introduction
There is renewed interest in nanographenes with nonalternant topologies owing to their role as chemical defects, such as Stone-Wales defect, in topological insulators, semiconductors, graphene quantum dots, and qubits elements.1 In particular, the introduction of well-defined defects into nanographenes is an effective strategy for tuning their physicochemical properties.2 Among the defects, the pentagon-heptagon pair (or azulene unit), which can be considered a minimum defect in graphene, has garnered considerable attention in recent years in studying the extraordinary chemical and physical properties of nanographenes.3, 4 In addition to incorporating non-hexagonal rings, introducing heteroatoms into the carbon skeleton is an effective strategy for further modulating the optical and electronic properties of nanographenes to achieve the desired functions.5, 6 For instance, the nitrogen (N) atom is an exceptional electron donor because its lone pair electrons can participate in the π-skeleton of nanographenes, generating interesting photophysical properties compared with those of the corresponding pure-carbon analog.5a, 5b, 6, 7, 8 In addition, incorporating N atoms into nonhexagon-embedded nanographenes could alter the molecular geometry into a curved conformation, which in turn would tailor not only the photophysical properties but also their supramolecular behavior.9 In 2021, Gryko et al. reported N-doped bowl-shaped nanographene 110 (Figure 1a) bearing joined pentagons and heptagons, which was achieved through multiple arylation steps. Using the same strategy, they also prepared larger π-conjugated nanographene 411 with two more heptagons. Recently, Kivala et al. synthesized an unexpected N-embedded nanographene 212 (Figure 1a) containing joined pentagon and heptagon via oxidative Scholl reaction. Recently, Lindner et al. synthesized N-embedded precursor 313 with a joined pentagon and heptagon, starting from the N-incorporated heptagon precursor through direct arylation (Figure 1a). Compound 3 could be further explored to produce a series of N-doped π-expanded nanographenes that exhibit intriguing thermally activated delayed fluorescence properties. Very recently, Tan et al. synthesized a larger N-doped nanographene 514 bearing six pentagon-heptagon pairs through photo-induced radical cyclization. In addition to these elegant molecular structures, substantial efforts have been dedicated to synthesizing novel N-doped nanographenes bearing nonhexagonal rings over the past few years. However, the development of methodologies for these highly desired molecules lags behind owing to the challenges of incorporating N atoms at specific positions.

Representative examples of nanographenes with nonhexagons. a) N-doped nanographenes with joined pentagon-heptagon pair(s). b) Nanographenes bearing heptagon obtained from the Pd-catalyzed reaction.
Over the past few decades, classic Scholl oxidative cyclodehydrogenation and direct arylation have been widely used to synthesize large π-systems, which have demonstrated significant potential for application in organic electronics.15, 16 However, both Scholl and direct arylation reactions involve unexpected rearrangements and tedious synthetic routes; therefore, the development of an effective synthetic strategy is highly desired. Recently, Würthner et al. reported a novel strategy for synthesizing nanographene 617 with heptagonal ring through Palladium (Pd)-catalyzed Carbon-Carbon coupling starting from borinic acid and 2,3-dibromonaphthalene (Figure 1b). Herein, we report a facile method to synthesize a series of nanographenes containing N-doped joined pentagons and heptagons (M1–M10, Figure 2). This method involves ring expansion and starts with the easily obtained precursor 8H-benzo[3,4][1,2]azaborinino[5,6,1-jk]carbazol-8-ol (M0, Figure 2) in which the heptagonal spacer can be further modulated. Notably, apart from the solo formation of heptagonal ring, the regulation derived from various substrates endowed the resultant nanographenes with tunable physicochemical properties. Moreover, two pairs of pentagonal-heptagonal ring-incorporated N-doped molecules (D1–D10, Figure 2) could also be obtained using the same strategy with excellent yield from precursor 5a,14a-diaza-5,14-diboradibenzo[a,m]rubicene-5,14-diol (D0, Figure 2). This synthetic approach can be further explored to build N-doped nanographenes with joined pentagons and octagons (ME1–ME3 and DE1–DE3, Figure 2), which have rarely been explored owing to the lack of a powerful synthetic approach,18 demonstrating the versatility and high efficiency of the proposed ring expansion method. Notably, the resultant nanographene bearing an anthracene-9,10-dione moiety (M9) exhibited tunable emission in a mixed solvent containing tetrahydrofuran (THF) and water (H2O) in which typical aggregation-caused quenching (ACQ) and aggregation-induced emission (AIE) behaviors were simultaneously observed in the same system, which is a quite rare phenomenon for graphene-type molecules.19

Synthetic strategy developed in this study.
Results and Discussion
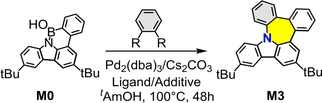
Borylating 3,6-di-tert-butyl-1-phenyl-9H-carbazole (compound 2 in Scheme S1) with boron tribromide (BBr3) followed by hydrolysis afforded the borinic acid precursor M0. Thereafter, precursor M0 was directly treated with 1,2-dibromobenzene in the presence of tris(dibenzylideneacetone)dipalladium (Pd2(dba)3) and dicyclohexyl(2′,6′-dimethoxy[1,1′-biphenyl]-2-yl)phosphane (SPhos), affording N-nanograhpene M3 with a pentagon-heptagon pair in a 1 % isolated yield (Table 1). Notably, the replacement of SPhos with tri-tert-butylphosphonium tetrafluoroborate (tBu3PHBF4) increased the yield to 18 % (Table 1). Moreover, the yields could be boosted from the starting material 1,2-dichlorobenzene (<1 %) to 1,2-diidobenzene (27 %) owing to the higher reactivity of the iodine group (Table 1).
|
||||
|
|
|
|
|
|---|---|---|---|---|
Entry |
R |
Ligand |
Additive |
Yield [%] |
1 |
Br |
PCy3 |
/ |
0 |
2 |
Br |
SPhos |
/ |
1 |
3 |
Br |
tBu3PHBF4 |
CsF |
5 |
4 |
Cl |
tBu3PHBF4 |
/ |
<1 |
5 |
Br |
tBu3PHBF4 |
/ |
18 |
6 |
I |
tBu3PHBF4 |
/ |
27 |
With the optimized reaction conditions in hand, we further evaluate the effect of electrons on the yields of resultant N-doped nanographenes. Electronic properties ranging from electron-deficient to electron-rich were introduced into the substrates accordingly (Table 2). First, the mono-products M1–M6 were synthesized from precursor M0 in various yields. Subsequently, the satisfactory results obtained using the new process encouraged us to explore nitrogen-doped precursor D0, which afforded di-products D1–D6 in acceptable yields (Table 2). Notably, the electron-deficient or electron-rich moieties did not have significant effect on the yields of the mono-products and di-products (M1–M6 and D1–D6, Table 2), suggesting the excellent tolerance of the proposed ring expansion method. Furthermore, the robustness of this synthetic method was demonstrated by treating aryborinic acid M0 with various dibromo aromatic substrates, affording nanograhpenes with N-doped pentagon-heptagon pairs (Table 3). For instance, the treatment of borinic acid M0 with 2,3-dibromoanthracene-9,10-dione afforded the corresponding product M9 in a 10 % yield. Notably, M9 exhibited both ACQ and AIE phenomena (see below). The approach adopted in this study furnished N,S-codoped nanographenes M8 and M10 in acceptable yields, indicating the possibility of post-functionalization with this class of compounds. Accordingly, the corresponding N-doped di-products D7–D10 bearing two pairs of pentagonal-heptagonal rings were obtained under similar conditions (Table 3). Notably, the yield of dual N-nanographenes was more compared to that of mono N-nanographenes in which the yield of D7 was up to 50 %.


The molecular structure of M10 bearing N-doped pentagon-heptagon pair was unambiguously revealed through single-crystal analysis.20 M10 adopted a curved geometry with dihedral angles of 39.42 and 48.86° (Figure 3a). Two different conformations of enantiomers P and M coexist at a ratio of 1 : 1 in the solid state, which were stabilized by π–π interactions with a minimum distance of 3.226 Å (Figure 3b). In the packing pattern, M10 crystallized along the b-axis of the unit cell with a pair of enantiomers (P and M) to afford a three-dimensional wavy structure (Figure 3c). Furthermore, di-product D3 containing two N-doped pentagon-heptagon pairs was also unequivocally confirmed through X-ray crystal analysis (Figures 3d and e).20 Compound D3 adopted a saddle-shaped geometry, with two opposite pairs of benzenoid rings alternating up and down the plane of the central 5[2,11]2-indolo[3,2-b]carbazole. The dihedral angles were determined as 51.01 and 56.56° in the solid-state owing to the large steric hindrance of the periphery rings (A−D). The enantiomers of P and M of D3 were arranged in a crisscross pattern with minimal distances of 3.549 and 3.466 Å, respectively, in the crystal packing (Figure S1a).

X-ray crystallographic molecular structures of M10 and D3. The hydrogen atoms are omitted for clarity. a) Top view and b) side view of M10. c) Crystal packing of M10. d) Top view and e) side view of D3. f) Bond lengths of azulene and N-doped pentagon-heptagon pairs in M10 and D3.
Temperature-dependent density functional theory (TD-DFT) calculations were performed at the B3LYP/6-31G(d) level of theory to determine the effect of different side substituents on the frontier molecular orbitals (Figures 4a and S95). The highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of M3 and D3 were delocalized over the entire molecule. However, the HOMOs and LUMOs of M9 and D9 were highly separated in which both the molecular orbital densities in the LUMO levels were primarily located on the anthracene-9,10-dione subunit, indicating effective tuning of the frontier molecular orbitals through different moieties. Accordingly, the HOMO–LUMO gaps decreased from M3 to D3 owing to the large π-conjugation. The anthracene-9,10-dione subunit, regarded as an electron-withdrawing group, reduced the HOMO–LUMO gap in M9 and D9 (Figure 4a). The aromaticity of the N-doped pentagon-heptagon pairs was investigated by nucleus-independent chemical shift (NICS) calculations performed at the GIAO-B3LYP/6-311+G(2d,p) level of theory (Table S6). The NICS values of representative examples of compounds M10 and D10 are shown in Figure 4b. The calculated NICS values for the heptagonal rings in compounds M10 and D10 were 13 and 10, respectively, indicating the antiaromatic properties compared to that of −5.5 in the pristine azulene. This result is also supported by their structural information; in the single crystal, the bond lengths of nitrogen-doped heptagon in M10 and D10 were measured from 1.398 to 1.484 Å, there is no longer an alternation of bond lengths compared to the heptagon in the pristine azulene (Figure 3f). The antiaromatic character of the heptagonal rings in M10 and D10 was further confirmed by the anti-clockwise ring current flow in the anisotropy of the induced current density (ACID) plot (Figure 4b and Figure S94). The aromatic properties of the carbazole moiety in compounds M10 and D10 were maintained according to the calculated NICS values.

a) Frontier molecular orbitals of representative compounds M3, M9, D3, and D9 obtained through TD-DFT at the B3LYP/6-31G (d) level. b) NICS(1)zz-avg values and calculated ACID plots of representative molecules M10 and D10.
The ultraviolet-visible (UV/Vis) absorption and photoluminescence (PL) spectra of mono-products M1–M10 and di-products D1–D10 were obtained in a dichloromethane (DCM) solution (Figure 5). The mono-products and di-products exhibited similar UV-vis absorption profiles. Owing to the extended π-conjugation, the peaks of maximum absorption and emission of the di-products were redshifted compared to those of the mono-products. In addition, the Stokes shifts of the di-products were substantially smaller (0.5 eV for D3; 0.45 eV for D7, and 0.45 eV for D8) than those of mono-compounds (0.96 eV for M3, 0.89 eV for M7, and 0.62 eV for M8), demonstrating that the di-products exhibit a more rigid structure. The di-compounds exhibited higher quantum yields (0.62 for D3, 0.52 for D7, and 0.31 for D8) compared to those of mono-compounds (0.26 for M3, 0.15 for M7, and 0.06 for M8). Notably, M9 exhibited both ACQ and AIE behavior (Figure 5e), which are rare phenomena for nanographenes.19 The fluorescence spectra of M9 exhibited an emission peak at 647 nm in a pure THF solution. After increasing the volumetric ratio of water, the emission intensity at 647 nm decreased, indicating typical aggregation-caused quenching. A new emission peak appeared at 627 nm when the water fraction exceeded 50 %. Furthermore, the fluorescence intensity significantly increased with higher water content, indicating a clear AIE phenomenon. These ACQ and AIE characteristics of M9 are likely due to the strong π-π stacking interactions between individual molecules in aggregation.19 The photophysical properties of the representative examples of di-products D1–D10 are summarized in Table 4 (for others, please refer to Table S5 in the Supporting Information).

a) UV/Vis absorption spectra and b) fluorescence spectra of M1–M10 in DCM. c) UV/Vis absorption spectra and d) fluorescence spectra of D1–D10 in DCM. e) Fluorescence spectra of M9 in THF/water mixtures with different water fractions (fw) obtained through irradiation at 450 nm. f) Cyclic voltammetry of representative compounds M3, M9, and M10 in DCM containing 0.1 M n-Bu4NPF6 at a scan rate of 50 mV s−1.
|
λabs[a] [nm] |
λem[a] [nm] |
ΔS[b] |
ϵabs[c] [×104] |
QY [%][d] |
LUMO[e] [eV] |
HOMO[f] [eV] |
ΔEgcal[g] [eV] |
ΔEgopt[h] [eV] |
|---|---|---|---|---|---|---|---|---|---|
D1 |
366 |
572 |
1.22 |
2.0 |
6 |
/ |
/ |
3.34 |
2.62 |
D2 |
417 |
499 |
0.49 |
1.2 |
50 |
−3.12 |
−5.75 |
3.37 |
2.63 |
D3 |
423 |
510 |
0.5 |
1.3 |
62 |
−3.09 |
−5.74 |
3.27 |
2.65 |
D4 |
430 |
510 |
0.45 |
1.1 |
60 |
−3.12 |
−5.67 |
3.3 |
2.55 |
D5 |
437 |
523 |
0.47 |
0.9 |
65 |
−3.1 |
−5.63 |
3.28 |
2.53 |
D6 |
435[i] |
518[i] |
0.46[i] |
0.5[i] |
58[i] |
−3.2[i] |
−5.73[j] |
3.33 |
2.53[i] |
D7 |
422 |
499 |
0.45 |
0.9 |
52 |
−3.09 |
−5.71 |
3.34 |
2.62 |
D8 |
432 |
516 |
0.47 |
2.0 |
31 |
−3.13 |
−5.68 |
3.3 |
2.55 |
D9 |
404 |
/ |
/ |
1.2 |
/ |
−3.71 |
−5.93 |
2.52 |
2.22 |
D10 |
415 |
626 |
1.01 |
2.3 |
11 |
−3.43 |
−5.96 |
2.97 |
2.53 |
DE1 |
373 |
527 |
0.97 |
2.0 |
10 |
/ |
/ |
3.27 |
3.08 |
DE2 |
355 |
/ |
/ |
4.4 |
/ |
/ |
/ |
3.26 |
3.04 |
DE3 |
411 |
523 |
0.65 |
2.8 |
7 % |
/ |
/ |
2.81 |
2.77 |
- [a] In dichloromethane solution; [b] ΔS=Stokes shift; [c] In dichloromethane solution; [d] The absolute QY via using the integrating sphere; [e] LUMO=HOMO+ΔEgopt; [f] determined by CV in dichloromethane solution; [g] Calculations were carried out at the CAM-B3LYP/6-31G+g(d) level; [h] ΔEgopt=optical band gap, ΔEg=1240/λabsonset; [i] in toluene solution; [j] determined by CV in orthdichlorobenzene.
The electrochemical properties of all the mono-products and di-products were subsequently investigated through cyclic voltammetry in a DCM solution (Figures S3a and S3b). Figure 5f shows the results of the representative examples of compounds M3, M9, and M10. No reduction behavior was observed for compound M3, whereas compounds M9 and M10 underwent reversible reduction at a half-wave potential of approximately −0.9 V vs. ferrocene (Fc)/ferrocene+ (Fc+) and −1.4 V vs. Fc/Fc+, respectively, indicating that highly tunable electrochemical properties were obtained by introducing different moieties on the heptagonal ring. Upon oxidation, compounds M3, M9, and M10 exhibited reversible behavior at a half-wave potential of approximately 1.3 V vs. Fc/Fc+, which is consistent with the calculated energy of the HOMO level.
In addition to the formation of N-doped pentagon-heptagon pairs, this ring expansion method has been further explored to synthesize N-doped nanographenes containing different types of nonhexagonal pairs, such as pentagon-octagon pairs, which have rarely been explored owing to the lack of a powerful synthetic approach. In particular, the formation of octagonal rings in nanographenes provides a foundation for realizing the well-known Mackay–Terrone crystals.21 By treating 1,8-dibromonaphthalene with precursor M0 under optimized conditions, compound ME1 was obtained in a 15 % yield through this effective method (Table 5). Furthermore, starting from 5,6-dibromoacenaphthylene, the extended product ME2 was synthesized in a 15 % yield, highlighting the efficiency of the proposed ring expansion method. Additionally, the octagonal ring was further functionalized with an imide group, generating ME3 in a 25 % yield from 6,7-dibromo-2-pentyl-1H-benzo[de]isoquinoline-1,3(2H)-dione. Therefore, di-compounds DE1, DE2, and DE3 were also synthesized from the precursor D0. Similar to the pentagon-heptagon pairs, the yield for di-compounds with pentagon-octagon pairs was significantly higher than that of mono-products (Table 5).

A single crystal of ME1 was obtained by slowly diffusing methanol into its solution in carbon disulfide. X-ray crystallographic analysis precisely demonstrated the presence of N-doped pentagon-octagon pairs in ME1 (Figure 6a and b).22 The highly twisted structure of ME1 was indicated by two large dihedral angles of 73.07 and 56.44°. In the packing pattern, compound ME1 crystalized along the b-axis with two pairs of enantiomers (P and M) alternately growing in opposite directions (Figure S1d). The bond length of the octagon in ME1 ranging from 1.389 to 1.501 Å suggested a decreasing aromaticity (Figure 6c), which was further confirmed by the calculated NICS value and the ACID plot (Table S6 and Figure S94). The frontier molecular orbital information was determined using TD-DFT at the B3LYP/6-31G(d) level of theory (Figures 6d and S95). Notably, the LUMO and HOMO distributions were separated for all the obtained N-doped nanographenes with pentagon-octagon pairs in which the molecular orbital density in the LUMO levels is primarily located on the naphthalene-based subunits, despite the different naphthalene-based moieties introduced through the proposed ring expansion method.

a) Top view and b) side view of the X-ray crystallographic molecular structure of ME1. c) Bond length of N-doped pentagon-octagon pair in ME1. d) LUMO and HOMO distributions of DE1–DE3.
The UV/Vis absorption and PL spectra of mono-products ME1–ME3 and di-products DE1–DE3 were measured in a DCM solution (Figures 7a and b). The peaks of maximum absorption of compound DE1 were significantly redshifted compared with those of compound ME1 in the presence of similarly shaped UV/Vis absorption spectra. Compounds ME2 and DE2 exhibited weak absorption in the range of 400–500 nm, resulting from the extra fused five-membered ring. The absorption peaks of ME3 and DE3 were observed at approximately 410 nm with emission peaks at 527 nm and 523 nm, respectively, resulting in a larger Stokes shift (0.68 eV for ME3, 0.65 eV for DE3) compared to the results obtained by Lindner (0.18 eV)7 and Würthner (0.09 eV)8 with N-doped six-membered ring, indicating a promising application in fluorescence imaging.23 The observation of low quantum yield in DE1 and DE3 (10 % for DE1 and 7 % for DE3) was probably due to the dynamic motion of N-doped cyclooctatetraene which accelerates the nonradiative decay process from the excited state.24, 26b Notably, a dual emission (DE) phenomenon was observed in compound ME3, which is likely due to the conformation flexibility resulting in two stable conformers.25 Normally, for cyclooctatetraene-embedded polycyclic aromatic hydrocarbons, excited state conjugation enhancement (ESCE) induces dual emission (DE).26 The relaxed long-wavelength state with extended conjugation is reached via a finite but thermally accessible barrier through the large bending motion of cyclooctatetraene.26a Therefore, a relaxed potential energy surface (PES) scan of ME3 at the PBE0/6-31G(d) level was performed to determine the energy difference between different bending motions. As shown in Figure 7c, along with the change in the dihedral angle in N-doped [4]hetero-helicene, the total energy increased and subsequently decreased. As a result, two stable conformers, ME3-geo1 and ME3-geo2, were confirmed. In addition, the energy difference between ME3-geo1 and ME3-geo2 was 2.61 kcal mol−1, indicating the existence of a high percentage of the conformer ME3-geo1 in compound ME3. In contrast, the highest energy barrier from ME3-geo1 to ME3-geo2 was 6.02 kcal mol−1, indicating fast isomerization even at room temperature (Figure 7c).

a) UV/Vis spectra and b) fluorescence spectra of ME1–ME3 and DE1–DE3 in DCM. c) Relaxed PES scan (opt=modredundant) of ME3 at the PBE0/6-31G(d) level. All calculations were corrected for dispersion (D3BJ). d) TD-DFT calculated maximum fluorescence wavelengths of ME3-geo1 and ME3-geo2 at the PBE0/6-31G(d) level. f value represents the oscillator strength. The SMD solvent effects with DCM were considered. All calculations were corrected for dispersion (D3BJ).
TD-DFT calculations were performed based on the two distinct conformers. As shown in Figure S96, the calculated maximum UV/Vis absorption peak of ME3-geo1 is located at 410 nm, which is consistent with the experimental UV/Vis data (408 nm). For ME3-geo2, the calculated maximum UV/Vis absorption peak was 487 nm, which is longer than that of ME3-geo1 (Figure S97), which may be caused by the extended conjugation structure. The non-observed absorption peak at approximately 487 nm in the experiment may be due to the low percentage of ME3-geo2 in compound ME3. Moreover, to identify the DE phenomena, the maximum fluorescence peaks of ME3-geo1 and ME3-geo2 were calculated using TD-DFT optimization (Figure 7d). Accordingly, the first and second experimental emission peaks exhibited at approximately 510 and 640 nm can be assigned to the emission peaks from ME3-geo1 (513 nm) and ME3-geo2 (645 nm), respectively, which can be attributed to the ESCE.26
Although the shapes and positions of the UV/Vis spectra for ME3 and DE3 were almost maintained in solvents with varying polarities (Figure 8a and b), significant solvatochromism was observed in the fluorescence spectra of ME3 and DE3 (Figure 8c and d). The PL peaks of ME3 redshifted from 481 to 529 nm with increasing solvent polarity. Similarly, a redshift from 474 to 541 nm was observed in compound DE3, indicating that large dipole moment changes exist at the excited state in different solvents, resulting in the occurrence of charge transfer excited states in ME3 and DE3.27 Accordingly, the photoluminescence quantum yield (PLQY) decreased with increasing solvent polarity (Table S6). These properties make such N-doped pentagon-octagon paired molecules viable candidates for sensor devices such as chemical probes.28

UV/Vis absorption spectra of a) ME3 and b) DE3 in different solvents; solvatochromic effect of c) ME3 and d) DE3 in solvents with varying polarities.
Conclusion
In conclusion, we developed a facile and effective strategy to synthesize a series of novel nanographenes bearing N-doped pentagon-heptagon pairs using a ring expansion method. The resultant chemical structures were undoubtedly determined through single-crystal X-ray analysis. By introducing various moieties on heptagons, the frontier molecular orbitals and their corresponding optoelectronic properties could be finely tuned, including AIE behavior. Notably, the efficiency of the proposed synthetic approach was further consolidated in the preparation of nanographenes containing N-doped pentagon-octagon pairs. The feasible synthetic method developed here could guide the preparation of novel N-doped graphene nanostructures bearing nonhexagonal pairs, such as graphene nanoribbons with N-doped nonhexagons, and provide various opportunities for tuning the structures and properties for future optoelectronic applications.
Acknowledgments
This work was supported by the Hong Kong Research Grants Council (27301720, 17304021), National Natural Science Foundation of China (22122114). J. L. is grateful for the funding from The University of Hong Kong (HKU) and ITC to the SKL. We thank the UGC funding administered by HKU for supporting the Time-of-Flight Mass Spectrometry Facilities under the Support for Interdisciplinary Research in Chemical Science. We acknowledge the computer cluster (HPC2021) of HKU for generous allocations of compute resources.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

