

Reactivity of Glutaconamides Within [2]Rotaxanes: Mechanical Bond Controlled Chemoselective Synthesis of Highly Reactive α-Ketoamides and their Light-Triggered Cyclization
Graphical Abstract
The mechanical interlocking of glutaconamides is crucial for their chemoselective oxidation to β,γ-unsaturated α-ketoamides. These unprecedented species were satisfactorily employed in blue-light-driven cyclizations to divergently provide hydroxy-2-azetidinones or oxazolidinones through two modes of the Norrish/Yang type-II reaction. This behavior starkly differs from that of the non-interlocked substrates under the same reaction conditions.

Abstract
Glutaconamide-based [2]rotaxanes are efficiently oxidized to the respective interlocked α-ketoamides, whereas their non-interlocked threads afford hydroxycyclohexene tetraamides under similar reaction conditions. These results showcase the mechanically interlocking of highly reactive substrates as a powerful tool for controlling their chemical behavior. Inside the macrocycle and under irradiation with light, the α-ketoamide threads convert, in a divergent manner, into the corresponding interlocked hydroxy-β-lactams or oxazolidinones by two modes of Norrish/Yang type-II intramolecular cyclizations, processes that are efficiently chemocontrolled by the mechanical bond.
Enzymes, considered the most powerful catalysts in nature, selectively control numerous biochemical processes.1 The active site, located in the inside of their three-dimensional structure, catalyzes processes with high specificity. Mechanically interlocked molecules, as prototypes of artificial molecular machines,2 have recently shown intriguing applications in synthesis by taking advantage of the preorganization of the entwined components, simulating enzymatic behavior.3 In the case of [2]rotaxanes, it is possible to design their molecular architecture,4 by modifying their interlocked components,5 in which the preservation of the mechanical bond is crucial.6 In addition, the chemical modification of the functionalities at the thread remains highly challenging due to the archetypal shielding protection of the macrocycle, which decreases their reactivity,7 but at the same time creates a confined space where a chemical process can be controlled.8 In this line, we recently found an unprecedented effect of the mechanical bond, which activates and diastereocontrols an intramolecular cyclization of the thread, protecting the resulting compounds, unstable under these reaction conditions (Figure 1a). Benzylfumaramide threads, forming part of [2]rotaxanes, reacted with a base affording interlocked trans-β-lactams.9 In contrast, the reactions of the non-interlocked threads yielded diastereomeric mixtures of lactams in low yield, along with decomposition products. Goldup and co-workers reported an unexpected tandem active template Cu-mediated alkyne-azide cycloaddition-rearrangement process during a rotaxane formation, which occurs only in the presence of a bipyridine macrocycle.10 Instead of the expected triazole derivatives, acrylamide-based rotaxanes were obtained (Figure 1b).

Effects of the mechanical bond in: a) the cyclization of interlocked and non-interlocked fumaramides;9 b) the Cu-mediated alkyne-azide cycloaddition (CuACC) in the presence or absence of a macrocycle;10 c) the base-promoted aerobic oxidation of interlocked and non-interlocked glutaconamides (this work).
Herein we explore the reactivity of [2]rotaxanes, with glutaconamide-base threads, a function scarcely found in bibliography,11 and how the mechanical bond influences the outcome of a transformation (Figure 1c). The reactions with the interlocked systems showed to be entirely different to that of the nude threads. Whereas the reaction of the glutaconamide threads with a base yielded cyclohexene-derived tetraamide derivatives, the rotaxanes exclusively provided interlocked α-ketoamides, obtained through a rare base-triggered aerobic α-oxidation,12 with the mechanical bond fully controlling the selectivity. Finally, we used the α-ketoamides as substrates in Norrish/Yang type-II cyclizations, chemoselectively yielding interlocked hydroxy-β-lactams and oxazolidinones.13
Initially, glutaconamide-based threads 1 were tested as templates for the obtention of hydrogen-bonded rotaxanes 2 (Scheme 1, see Supporting Information for further details). The reaction of tetrabenzylglutaconamide E-1 a with p-xylylenediamine and isophthaloyl chloride in the presence of Et3N yielded a mixture of the two geometrical isomers of rotaxane 2 a in a 6 % combined yield (E : Z ratio of 45 : 55). Unreacted thread 1 a was recovered (88 %) as a mixture of isomers (E : Z ratio, 86 : 14). This scenario was general for the synthesis of six rotaxanes 2, having different bulky groups at the ends (see Supporting Information for further details). The presence of an additional methylene in these systems drastically decreased the yields of the rotaxane formation reactions when compared to the fumaramide-based threads previously reported.9

Synthesis of rotaxanes 2. Reagents and conditions: (i) p-xylylenediamine, aroyl dichloride, Et3N, CHCl3, 25 °C, 4 h. Inset A: X-ray structure of E-2 a. Intramolecular HB lengths [Å] (and angles [°]): N5 H05⋅⋅⋅O1 2.06 (154); N4 H04⋅⋅⋅O2 2.14 (161); Inset B: X-Ray structure of Z-2 a: Intramolecular HB lengths [Å] (and angles [°]): N3 H03⋅⋅⋅O1 2.11 (169); N5 H05⋅⋅⋅O2 2.09 (176); N6 H06⋅⋅⋅O2 2.09 (174). For clarity, selected hydrogen atoms and solvent molecules are omitted.
Since E/Z mixtures of rotaxanes 2 were obtained from pure (E)-threads 1, we decided to follow by 1H NMR the isomerization of thread 1 a and rotaxane 2 a in CDCl3 at 25 °C in the presence of Et3N (see Supporting Information for further details). Threads E-1 a and Z-1 a equilibrated till reaching the same isomeric ratio after 2 h (E : Z, 86 : 14) (Figures S4 and S5). Moreover, the addition of D2O triggered the formation of the deuterated thread 1 a–d4 (98 % D, E : Z ratio, 86 : 14) (Figure S10–11), indicative of the intermediacy of a delocalized allylic anion. In contrast, neither isomerization nor deuteration occurred in the case of the rotaxanes E-2 a or Z-2 a, showing the enhanced stability of their threads due to the presence of the sterically hindered macrocycle, which precludes the approaching of the base (Figure S6).14 These data verified that isomerization of starting thread 1 a in the rotaxanation reaction should occur before the formation of the rotaxanes. The predominance of Z isomers in rotaxanes 2 a–e (equal ratio in 2 f), despite Z-threads are the minor isomers at the beginning of the reaction, indicated the better templating ability of these latter isomers. The hydrogen bonding network established between the thread and the macrocycle (or intermediates) in Z-2 a and E-2 a might be the reason behind their different templating ability. Indeed, the solid structure of both isomeric rotaxanes 2 a, elucidated by SC-XRD, showed that the macrocycle (in boat conformation) in Z-2 a established three hydrogen bonds (HB) with the embedded thread, including one stabilizing bifurcated HB, whereas in E-2 a (macrocycle in chair conformation) only two HBs were observed (Scheme 1, Insets A–B).15
Considering our previous studies on the CsOH-promoted cyclization of interlocked fumaramides for the selective formation of β-lactams,9 we envisioned that glutaconamide-based systems 2 (with one extra carbon at the thread) could react similarly. However, the reaction of 2 a with CsOH in DMF gave an irresoluble complex mixture of products. Remarkably, the election of K3PO4 as a milder base (see Supporting Information for the screening of the reaction conditions, Tables S1–S3) allowed us to isolate the interlocked α-ketoamide 4 a (E isomer, elucidated by SC-XRD, Scheme 2a, Inset A) as the main product, resulting from an allylic oxidation process at the thread.16, 17 We followed this process by 1H NMR, with a solution of 2 a in DMF-d7 at 25 °C under open air and in the presence of K3PO4, first observing a fast Z to E isomerization, followed by the allylic oxidation. After 4 h, the starting material was fully converted into a mixture of oxidized rotaxane 4 a and free macrocycle (ratio 4 a: macrocycle, 60 : 40), probably as result of the instability of 4 a in basic media (Figure S1).18 In stark contrast, the reaction of non-interlocked 1 a under the same conditions yielded the cyclohexene-derived tetraamide 3 a instead of the expected α-oxoamide 5 a (Scheme 2b, Inset B).19 These results clearly show that the divergent reactivity of the oxoamide is controlled by the presence or absence of the mechanical bond. The formation of products 3 and 4 can be mechanistically explained (Scheme 2c, see also Figures S19 and S20) as starting by the deprotonation of the acidic allylic position of the thread in 1 or 2 affording anion A, which reacts with adventitious oxygen yielding peroxyanion B.20 The release of a hydroxyl anion results in the formation of ketoamides 4 and 5. In the absence of the mechanical bond, ketoamide 5 undergoes conjugate addition of anion A to give intermediate C (isolated in the reaction of thread 1 d, see Supporting Information). Finally, deprotonation of C followed by diastereoselective nucleophilic attack of the formed anion D over the keto group affords compound 3 (isolated for threads 1 a,c–d, see Supporting Information). On the basis of this mechanism, a base-catalyzed process should be feasible. Indeed, in the presence of a sub-stoichiometric amount of base (20 mol %), full conversion of thread 1 a was achieved after two days (Table S4). With rotaxane 2 a, the reaction stopped at 30 % conversion because of the appearance of free macrocycle, which quenched the catalytic process by reacting with the base (Table S3).21 Importantly the reaction performed in deoxygenated DMF and under nitrogen atmosphere did not proceed (0 % conversion), an indication of the needing of molecular oxygen as the oxidant. In parallel, we tested other conditions for this allylic oxidation, such as the use of SeO2 as an oxidant.22 Heating a solution of Z-2 a with an excess of SeO2 in dioxane at 90 °C for 48 hours afforded 65 % yield of 4 a, together with byproducts derived from thread decomposition, such as dibenzylformamide or dibenzylamine. Interestingly, under the same conditions, the reaction of thread E-1 a gave a low yield of oxidized thread 5 a (10 %) along with high amounts of byproducts (threads 1 c and 1 d were also tested under these conditions, obtaining low yields of the oxidized 5, see Supporting Information for further details).23 These results highlight the shielding effect of the macrocycle, protecting the α-ketoamide thread of 4 a against overoxidation reactions.

Reaction of: a) rotaxane 2 a; and b) thread 1 a in the presence of K3PO4. Inset A: X-ray structure of E-4 a. Intramolecular HB lengths [Å] (and angles [°]): N3 H03⋅⋅⋅O3 2.95 (154.1); N4 H04⋅⋅⋅O1 2.19 (138.9); N5 H05⋅⋅⋅O2 2.28 (167.1). Inset B: X-ray structure of 3 a. For clarity, selected hydrogen atoms and solvent molecules are omitted; c) plausible mechanism for the synthesis of 3 and 4. Reagents and conditions: K3PO4 (2 equiv), DMF, 25 °C. [a] Yield of E-4 a obtained by reaction of 2 a with SeO2 (50 equiv) in dioxane at 90 °C for 48 h; [b] Yield of E-5 a obtained by reaction of 1 a with SeO2 (5 equiv) in dioxane at 90 °C for 24 hours. The dashed blue macrocycle indicates the presence or absence of the macrocycle.
The remaining rotaxanes 2 were tested in the oxidation reaction under the two established conditions, in the presence of K3PO4 (Method A) or by the action of SeO2 (Method B) (Scheme 3).24 The reaction of rotaxane 2 b (with NO2 groups in the macrocycle) gave similar results to those of 2 a, indicating that the presence of electron-withdrawing groups in the macrocycle has a negligible effect on the process. In contrast, the structural variation of the substituents at the N atoms of the amide function of the threads did influence the reactivity. While rotaxane 2 c (with p- methoxybenzyl groups) yielded the oxidized compound 4 c in a similar yield as 2 a, the oxidation of rotaxane 2 d (phenyl and benzyl groups) gave 4 d in almost quantitative yield (95 %) in the reaction with SeO2, showing a higher stability against overoxidation processes. Rotaxane 2 e, with smaller stoppers (n-butyl chain), was unstable under heating with SeO2 (the competitive dethreading process was observed), while in the presence of base, we only observed decomposition byproducts. Finally, isobutyl-substituted rotaxane 2 f was unreactive under both conditions.

Oxidation of rotaxanes 2. Reagents and conditions: i) Method A: K3PO4 (2 equiv), DMF, 25 °C, open air, 4 h; Method B: SeO2 (50 equiv), dioxane, 90 °C, 48 h. [a] 2 e dethreaded at this temperature. [b] No reaction took place. In parentheses, yields using method B.
α-Oxoamide derivatives are known to undergo Norrish/Yang type-II cyclizations under light irradiation, yielding oxazolidinones or hydroxy-β-lactams as the main products, depending on the reaction conditions.13 Thus, we irradiated solutions of rotaxanes 4 a–d in CH2Cl2 under blue light in the presence or absence of silica gel,25 thus observing notable differences in the chemoselectivity of the process (Table 1, see Figure S2). The irradiation of a solution of rotaxane 4 a for 1 h triggered the formation of the interlocked hydroxy-β-lactam 6 a as the main product (cis isomer, elucidated by SC-XRD,26 Table 1, Inset A) together with a minor amount of oxazolidinone 7 a.27 In contrast, the irradiation of a solution of 4 a with suspended silica gel yielded the oxazolidinone 7 a (structure elucidated by SC-XRD,28 Table 1, Inset B) as the main product along with hydroxy-β-lactam 6 a as a minor component. This is a general behavior for the rest of rotaxanes 4 b–c. For rotaxane 4 d, with a phenyl group as the stopper, longer reaction times (48 h) were required to achieve full conversion. It is worth noting that irradiation of the nude thread 5 a mainly yielded the corresponding hydroxy-β-lactam, regardless of which of the two reaction conditions was employed (see Supporting Information for further details, Figure S3). These results show that the chemoselectivity of this cyclization process is neatly enhanced by the mechanical bond.
|
||||
Entry |
Rotaxane |
Method |
Ratio 6 : 7[b] |
Yield [%][c] |
|---|---|---|---|---|
1 |
4 a |
A |
72 : 28 |
25 |
2 |
B |
31 : 69 |
51 |
|
3 |
4 b |
A |
72 : 28 |
18 |
4 |
B |
30 : 70 |
45 |
|
5 |
4 c |
A |
75 : 25 |
19 |
6 |
B |
30 : 70 |
72 |
|
7 |
4 d[d] |
A |
55 : 45 |
19 |
8 |
B |
45 : 55 |
48 |
|
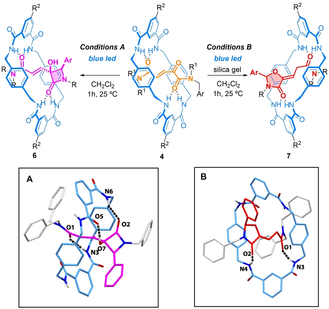
- [a] Reaction conditions: 4 (0.02 mmol) in CH2Cl2 (10 mL) (Conditions A) or in the presence of silica (100 mg) (Conditions B), blue LED, N2. Inset A: X-ray structure of 6 a. Intramolecular HB lengths [Å] (and angles [°]): O7 H07⋅⋅⋅O5 1.81 (166.8); N3 H03⋅⋅⋅O1 2.13 (167); N6 H06⋅⋅⋅O2 2.03 (163). Inset B: X-ray structure of 7 a. Intramolecular HB lengths [Å] (and angles [°]): N3 H03⋅⋅⋅O1 2.09 (159); N4 H04⋅⋅⋅O2 2.11 (173). For clarity, selected hydrogen atoms were omitted; [b] Determined by 1H NMR spectroscopy. [c] Combined yield of the isolated product. [d] 48 h were required.
In conclusion, the results reported herein demonstrate a set of appealing effects of the mechanical bond. First, it controls the outcome of an unusual base-triggered aerobic oxidation of interlocked glutaconamides. Whereas the free threads yielded polysubstituted cyclohexenes, the respective rotaxanes selectively afforded interlocked α-ketoamides. Using SeO2 as oxidant, the shielding effect of the mechanical bond protects the interlocked α-ketoamides against over-oxidation processes, whereas the non-interlocked ketoamides rapidly decompose. Finally, the chemoselectivity of the light-triggered intramolecular cyclization of the interlocked ketoamides is enhanced by the mechanical bond, affording interlocked hydroxy-β-lactams or oxazolidinones. This research enforces the role of the mechanical bond in [2]rotaxanes, influencing the reactivity in confined spaces such as a molecular flask, thus facilitating unusual chemical modifications and controlling their selectivity.
Acknowledgments
This work was supported by the Spanish Ministry of Science and Innovation (project PID2020-113686GB-I00/MICINN/AEI/10.13039/501100011033) and the Fundacion Seneca-CARM (project 21907/PI/22).
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article. CCDC 2241648, 2241650–2241654 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing [email protected].

