

Photocatalytic Arylation of P4 and PH3: Reaction Development Through Mechanistic Insight
Dedicated to Professor Holger Braunschweig on the occasion of his 60th birthday
Graphical Abstract
31P{1H} NMR spectroscopic studies provide significant, new insights into the mechanism of the recently reported photocatalytic arylation of white phosphorus (P4). Through the first-time observation of a series of reaction intermediates, these studies have also inspired the development of the first examples of direct, catalytic arylation of PH3, which is a key synthetic intermediate for industrial phosphorus chemistry.

Abstract
Detailed 31P{1H} NMR spectroscopic investigations provide deeper insight into the complex, multi-step mechanisms involved in the recently reported photocatalytic arylation of white phosphorus (P4). Specifically, these studies have identified a number of previously unrecognized side products, which arise from an unexpected non-innocent behavior of the commonly employed terminal reductant Et3N. The different rate of formation of these products explains discrepancies in the performance of the two most effective catalysts, [Ir(dtbbpy)(ppy)2][PF6] (dtbbpy=4,4′-di-tert-butyl-2,2′-bipyridine) and 3DPAFIPN. Inspired by the observation of PH3 as a minor intermediate, we have developed the first catalytic procedure for the arylation of this key industrial compound. Similar to P4 arylation, this method affords valuable triarylphosphines or tetraarylphosphonium salts depending on the steric profile of the aryl substituents.
Introduction
White phosphorus (P4) is the common precursor from which all organophosphorus compounds (OPCs) are currently prepared.1 The enormous range of important applications of these compounds (including pharmaceuticals, agrochemicals, photoinitiators, catalysts, et cetera) makes P4 one of the most important single precursors for the modern chemical industry.2 Unfortunately, current methods for the use of elemental phosphorus suffer from major drawbacks, including difficulties in the initial preparation of P4 from phosphate minerals as well as problems relating to phosphorus recovery and recycling.3 Particularly significant are serious limitations in state-of-the-art methods for the transformation of P4 into useful OPCs, which rely on wasteful and inefficient multi-step procedures involving extremely hazardous and difficult-to-handle reagents and intermediates.4 These problems are exemplified in the syntheses of arylated phosphines (Ar3P) and phosphonium salts (Ar4P+), which find important commercial applications in catalysis (among many other areas) and whose preparation involves such compounds as Cl2, PCl3 and molten Na (Figure 1 a).4, 5

a) Indirect, stepwise methods for the synthesis of triarylphosphines Ar3P and tetraarylphosphoniums Ar4P+ employed industrially; b) recently reported direct photocatalytic transformation of P4 into Ar3P and Ar4P+ and c) direct photocatalytic arylation of PH3 described herein, developed based on improved mechanistic understanding of P4 arylation.9
These deficiencies have prompted great interest in the development of alternative methodologies for the transformation of P4,6 and particularly those that have the potential to furnish OPCs directly in a single reaction, without the need for laborious isolation and manipulation of potentially hazardous intermediates.7 Unfortunately, despite extensive studies leading to numerous fundamental insights, the development of such new reactions has proven exceedingly challenging. In particular, while P4 is well known for being chemically reactive, researchers have struggled to find reactions that can selectively produce individual, useful OPCs in good yield.8
Recently, we reported a breakthrough in this area. We found that photoredox methods could be employed to successfully combine P4 with aryl iodides (ArI) to generate the corresponding Ar3P and Ar4P+ products under very mild conditions, often with markedly high selectivity (Figure 1 b).9 This is the first example of such a catalytic procedure furnishing these products directly from P4. Given the exponential growth in photoredox techniques reported over the past decade,10 including for P−C bond formation in other contexts,11 photoredox catalysis may also hold a much broader potential for the productive transformation of P4.12 Nevertheless, if this goal is to be successfully realised it is crucial to develop a deeper understanding of the mechanisms that underpin the existing photoredox reactivity of P4.13
Herein we report a detailed NMR spectroscopic study on the mechanism of photoredox-catalytic P4 arylation. From the results, we have been able to draw a number of significant conclusions. These include a recognition of the importance of side reactions in determining overall product yields, a rationalisation for the differing activities of precious metal and organic photocatalysts, and the identification of a viable minor reaction pathway that involves PH3 as an early reaction intermediate. Building upon the last of these, we have been able to develop the first examples of catalytic arylation of PH3, so providing an entirely new route to produce Ar3P and Ar4P+ products starting from this industrially important synthetic intermediate (Figure 1 c).
Results and Discussion
Previous studies and open mechanistic questions
As part of our initial report of photoredox-catalysed P4 arylation we described a series of preliminary mechanistic studies, on the basis of which we were able to propose an approximate outline mechanism for the catalytic phenylation of P4 to Ph4P+.9a This is reproduced in Scheme 1 a, and involves a fairly typical photoredox catalytic cycle for the generation of phenyl radicals (Ph. for Ar=Ph), in which the photoredox catalyst [1]PF6 (1=Ir(dtbbpy)(ppy)2, dtbbpy=4,4′-di-tert-butyl-2,2′-bipyridine, ppy=2-(2-pyridyl)phenyl; structure shown in Scheme 1) undergoes photoexcitation by blue light, followed by reductive quenching by Et3N (which acts as terminal reductant in this reaction), and oxidative regeneration by the substrate PhI. This last redox step prompts mesolytic fragmentation of the aryl halide substrate, generating radicals Ph.. These can then add to P4 to generate in sequence PhPH2, Ph2PH, Ph3P and Ph4P+, with the formation of these intermediates being observable by 31P{1H} NMR spectroscopy. The H atoms required for formation of PhPH2 and Ph2PH were proposed to derive from the reductant Et3N, presumably via direct abstraction of H. (HAT) or H+ following oxidation to Et3N.+. As part of the study described herein it was possible to confirm this hypothesis through use of deuterium-labelled Et3N-d15, which led to clear deuterium incorporation into these intermediates (see Section 3.6 of the Supporting Information for further details). Subsequent loss of these H atoms was proposed to proceed by formation of ArH (observed as a byproduct by 1H and 2H NMR spectroscopy).14

a) Outline mechanism for photocatalytic P4 arylation proposed previously; and b) recently reported photocatalytic P-H arylation of PhPH2 and Ph2PH.
Each of the individual arylation steps was previously confirmed to be photocatalytic. Indeed, in a recent follow-up study, we reported that the same methodology could be optimised to use PhPH2 and Ph2PH as starting materials for the generation of unsymmetrically substituted products (e.g. PhPAr3+, Ph2PAr2+; Scheme 1 b).9b This report also showed that the previously used precious metal catalyst [1]PF6 could be replaced by the inexpensive and easily accessible organic catalyst 3DPAFIPN (2; structure shown in Scheme 1) for the arylation of both P4 and PhPH2/Ph2PH.15
Despite the superficial simplicity of the catalytic cycle shown in Scheme 1 a it is clear that the overall mechanism is highly complex (we have emphasised previously that exhaustive phenylation of one equivalent of P4 requires the cleavage of six separate P-P bonds, formation of 16 new C-P bonds, and consumption of at least 16 equivalents each of PhI and Et3N) and that several important questions remain to be answered. From among these, at the outset of this study we identified three that are of particular significance:
1) Is it possible to identify any other intermediate(s) formed prior to PhPH2? Although PhPH2 is the first P-containing intermediate that it has been possible to observe during the phenylation of P4 thus far, it clearly cannot be the first such intermediate. Notably, by identifying PhPH2 and Ph2PH as intermediates it has been possible to develop new functionalisation reactions of these substrates,9b which might also be true for any earlier intermediates.
2) Is it possible to account for missing 31P intensity in these reactions? Preliminary in situ 31P{1H} NMR monitoring has shown that initial consumption of P4 is far faster than the formation of the first observable P-containing intermediate (PhPH2). Similarly, standard 31P{1H} spectroscopic measurements of the final reaction mixtures suggest that formation of the target products (e.g. Ar3P, Ar4P+) is typically very clean (no other P-containing species present, including P4), yet conversion to these products is consistently less than quantitative based on the initial amount of P4.
3) Can discrepancies in performance between catalysts [1]PF6 and 2 be explained? Although their photoredox properties are very similar, [1]PF6 has been found to give consistently higher product conversions for P4 arylation than 2. Conversely, 2 has been found to provide better conversions than [1]PF6 for analogous arylations starting from PhPH2/Ph2PH.
Identifying new intermediates and side-products during the arylation of P4
Having previously shown 31P{1H} NMR monitoring to be a valuable tool for the investigation of the P4 arylation reaction,9a we reasoned that answers to our question set might be found through more detailed NMR spectroscopic analysis. For consistency with previous studies, arylation using PhI was again chosen as the simplest model system. To begin, the phenylation of P4 using organic photocatalyst 2 was monitored over time, to provide a comparison with the analogous experiment already performed using [1]PF6 as catalyst.9a, 16 As expected, a similar overall reaction profile was observed (Figure 2 a), with rapid consumption of P4 followed by intermediate formation of PhPH2, Ph2PH and Ph3P, and ultimately formation of Ph4P+ as the major reaction product. Observation of a photo-CIDNP effect in the corresponding 1H NMR spectra was also consistent with previous observations using [1]PF6 (see Section 3.8 of the Supporting Information for more details).9a, 17 Nevertheless, some qualitative differences between the two catalysts could clearly be resolved. In particular, formation of the early intermediates PhPH2 and Ph2PH appears to peak significantly more quickly using 2, while the magnitude of these peaks is reduced (c.f. Figure 2 a,b).

Reaction profiles showing the evolution of P atom speciation during the light-driven phenylation of P4 (0.1 mmol scale) catalysed by 2 (a) or [1]PF6 (b), as assessed by time-resolved 31P{1H} NMR spectroscopy. Equivalents of reagent (equiv) and catalyst loadings (in mol %) are defined per P atom. Σ(unprod.) indicates the sum of intensities for unproductive side-products Side1–Side5. For a more comprehensive display of the kinetics see Section 3.9.1 in the Supporting Information.
More dramatically, in addition to the expected intermediates PhxPH3−x (x=1–3), 31P{1H} NMR monitoring clearly showed the formation of a significant number of other minor peaks during the reaction catalysed by 2 (for representative example spectra, see Figure 3 a). Some of these (designated Int1–Int4) were identified as intermediate species (either for productive or unproductive processes) based upon their concentration profile over time, while others (designated Side1–Side5) were identified as side-products and were persistent until the end of the reaction (Figure 2 a). The same side-products (Side1–Side5) could also subsequently be detected in the reaction catalysed by [1]PF6, but at appreciably lower concentrations (Figure 2 b). Nevertheless, the combined intensity of these signals accounts for a significant proportion of the overall 31P intensity in the final reaction spectra, and it seems feasible that there may be further unresolved signals of lower intensity, which could sum to a further significant value (vide infra).

Example 31P{1H} NMR spectra showing the formation of minor intermediates (Int1–Int4) and side-products (Side1–Side5) during the light-driven phenylation of P4 (0.1 mmol scale) catalysed by organocatalyst 2 (a); and tentatively proposed structures of these species based on in situ NMR data (see Sections 3.10 and 3.11 of the Supporting Information for more information) (b).
These observations suggest a clear explanation for less-than-quantitative target product formation highlighted above, with maximum formation of the final target products being limited by the formation of a collection of minor side products in amounts that are individually insignificant, but collectively substantial. Moreover, the fact that far greater amounts of these side-products are observed when using the organocatalyst 2 provides a rationale for the observation that 2 furnishes consistently poorer final product conversions for the arylation of P4, despite being a more productive catalyst for the optimised synthetic arylations of the “downstream” intermediates PhPH2 and Ph2PH (for a discussion of catalyst performance in these latter reactions see Section 3.9.3 of the Supporting Information).
Plausible structures of the species Int1–Int3 and Side1–Side5 are shown in Figure 3 b and are based upon detailed in situ NMR studies including 2D techniques and proton-coupled 31P spectra which are presented in the Supporting Information (Sections 3.10 and 3.11), alongside discussion of possible mechanisms of formation. As an illustrative example, the product Side1 could be assigned based on a combination of its 31P chemical shift (suggestive of a Ph2PNR2 species), long range 1H-31P correlation experiments (showing correlations between the 31P resonance and 1H signals at chemical shifts consistent with Ph and CH2Me peaks), and comparison with literature data.
Interestingly, the majority of these species appear to be derived from non-innocent behaviour of the Et3N terminal reductant (with the notable exception of Int3, Ph4P2, which was also observed in our previous investigation of the 2-catalysed reaction of Ph2PH with cyclohexyl iodide, and was proposed to arise through dimerization of transiently-formed radicals Ph2P.).9b Given that Et3N is widely used as a “privileged” terminal reductant in photoredox transformations, and that [1]PF6 and 2 have also been used to catalyse many other photoredox reactions, these results suggest that similar patterns of non-selective side-reactivity could be an underappreciated but highly significant limiting factor in many other photoredox transformations (indeed, related non-innocent side reactions have previously been highlighted in some other catalytic reactions).18 In the absence of the convenient 31P NMR handle, these would likely be extremely challenging to detect. Minimisation of these side reactions is a clear avenue for further reaction optimisation.19
To try and identify further reaction intermediates—particularly those that may form in the early stages of the reaction—additional monitoring experiments were carried out on reactions performed at lower temperatures and using higher catalyst loadings. It was anticipated that transient, early intermediates might be better able to accumulate under these conditions and, indeed, several further 31P{1H} resonances could be observed (see Figure 4 a; designated Int5–Int8; again, these may be intermediates for either productive or unproductive processes), and plausible corresponding chemical structures assigned are depicted in Figure 4 b (see also Section 3.11.1 of the Supporting Information). The proposed structures of Int5–Int7 further emphasise the non-innocent behaviour of the Et3N reductant.

a) 31P{1H} NMR spectrum showing the formation of further intermediates (Int5-Int8) of the light-driven phenylation of P4 (0.1 mmol scale) catalysed by [1]PF6. The spectrum was recorded in the dark after an illumination period of 30 min at 273 K (see also Section 3.11.1 of the Supporting Information). b) Proposed structures of Int5–Int8 observed during the light-driven phenylation of P4 catalysed by [1]PF6 and 2 at low temperatures and high catalyst loadings.
PH3 as a substrate for photocatalytic arylation
The specific observation of PH3 as one of the above intermediate signals (Int8; readily assigned on the basis of its characteristic chemical shift and binomial quartet splitting in the proton-coupled 31P spectrum, see Figure 4 a) was particularly intriguing.3b Although our previous mechanistic proposals (e.g. Scheme 1 a) had assumed PhPH2 to be the first P1 intermediate formed, this result—combined with our previous observation that PH3 can be formed upon irradiation of a combination of P4, Et3N and [1]PF6 in the absence of ArI—suggested an alternative synthetic possibility. Specifically, it was queried whether PH3 could in fact be a key intermediate formed prior to arylation: that is, whether arylation of P4 could proceed via initial Et3N-mediated reduction to PH3,20 with arylation only subsequently occurring. Notably, such a possibility would have broad implications beyond the immediate mechanistic significance. PH3 is a major synthetic intermediate, employed industrially for the preparation of many important phosphine and phosphonium derivatives. However, these syntheses proceed exclusively through the hydrophosphination of unsaturated substrates, which limits the scope of accessible products and thereby excludes the formation of aryl-substituted derivatives (Ar3P, Ar4P+). Indeed, efficient, direct arylation of PH3 remains a long-unmet synthetic goal that, though related, is distinct from the target of direct P4 functionalisation.3c, 4, 21
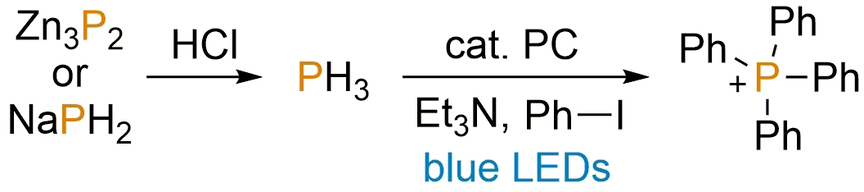
To this end, it was decided to investigate the deliberate use of PH3 as a substrate for our photoredox-catalysed arylation methodology. For reasons of practical simplicity, PH3 was not employed directly, but rather generated in situ using a two-chambered apparatus, inspired by a recent report by Ball et al. (Figure 5).22 Addition of aqueous HCl to solid Zn3P2 in one chamber liberates gaseous PH3, which can diffuse into the second chamber. This chamber contains the remaining components of the photocatalytic reaction mixture and is stirred under continuous blue LED irradiation (with concomitant water cooling to maintain approximately ambient temperature).

Schematic representation of the two-chambered apparatus used to investigate the photocatalytic phenylation of PH3, generated by in situ acidification of suitable solid precursors.
Gratifyingly, initial experiments using catalytic [1]PF6 and reaction conditions identical to those used previously in the optimised phenylation of P4 unambiguously confirmed the formation of Ph4P+ in modest yields (Table 1, entry 1). This result clearly illustrates the feasibility of PH3 as an intermediate en route to arylated products and to our knowledge is the first example of the successful catalytic arylation of PH3. Control experiments confirmed that all reaction components ([1]PF6, Et3N, light) were necessary to achieve appreciable product formation (Table S1, SI). Catalytic formation of Ph4P+ could also be achieved using organocatalyst 2 instead of [1]PF6 (Table 1, entry 2), illustrating that this reactivity is not dependent on the use of a precious metal, and is mechanistically feasible for an organocatalysed reaction.
Entry[a] |
PH3 source (mmol) |
PC (mol %) |
t [h] |
Solvent |
Conversion to Ph4P+ [%] |
|---|---|---|---|---|---|
1 |
Zn3P2 (0.05) |
[1]PF6 (2) |
18 |
MeCN/PhH (3:1) |
17 |
2 |
Zn3P2 (0.15) |
2 (10) |
48 |
MeCN/PhH (3:1) |
23 |
3 |
Zn3P2 (0.15) |
[1]PF6 (2) |
48 |
MeCN |
30 |
4[b] |
Zn3P2 (0.15) |
[1]PF6 (2) |
48 |
MeCN |
35 |
5[b] |
NaPH2 (0.1) |
[1]PF6 (2) |
24 |
MeCN |
41 |
6[c] |
NaPH2 (0.1) |
[1]PF6 (2) |
24 |
MeCN |
29 |
7[c,d] |
NaPH2 (0.1) |
[1]PF6 (2) |
24 |
MeCN |
68 |
- [a] Reactions performed using a 20 mL two-chambered apparatus described in Figure 5, using 12.5 equiv HCl, 11 equiv PhI, 14.4 equiv Et3N in 2.0 mL solvent under blue LED irradiation and N2 atmosphere. [b] A 10 mL two-chambered apparatus was used. [c] A 10 mL single-chambered apparatus was used. [d] No HCl was added. Equivalents of reagent (equiv) and catalyst loadings (in mol %) are defined per P atom.
Conversion to the target Ph4P+ could be significantly increased through modification of the reaction time, solvent, and concentration (Table 1, entry 3 and Table S2, SI). Although these conversions are still appreciably lower than those obtained previously when starting from P4, this may be attributable at least in part to differences in experimental setup. In particular, the need for gaseous PH3 to diffuse between reaction chambers before dissolving in the reaction solution is expected to significantly impact the rate of reaction. This conclusion was supported by reactions performed using a two-chamber apparatus of reduced volume, which led to a further improvement in performance (Table 1, entry 4; for full details of the apparatus see Section 2.1 of the Supporting Information). An additional improvement was observed upon replacement of Zn3P2 with NaPH2, as an alternative source of PH3 upon acidification (Table 1, entry 5).23
To further mitigate problems associated with gas transfer of PH3, the use of a single-chambered experimental apparatus was pursued, within which it was hoped that addition of acid would trigger release of PH3 directly into the photocatalytic reaction solution (for full details see Section 2.2.1 of the Supporting Information). Disappointingly, use of this apparatus in combination with NaPH2/HCl as a source of PH3 did not lead to an improvement in reaction outcome, although clear catalytic formation of Ph4P+ was once again observed (Table 1, entry 6). Gratifyingly, however, when this reaction was repeated in the absence of HCl significantly improved results were observed, with very good conversion to the target product achieved following slight further optimisation (Table 1, entry 7 and Table S3, SI). Thus, the conjugate base of PH3 appears to be a highly effective and convenient synthetic surrogate for PH3 in these reactions, allowing catalytic arylation to be performed in a practically convenient manner while achieving synthetically relevant conversion and selectivity.
Having identified an optimised set of reaction conditions for the [1]PF6-catalysed phenylation of PH3, the use of other, substituted aryl iodides was investigated (Table 2). Satisfyingly, a range of other substrates ArI could be employed successfully, including examples bearing both electron-donating and electron-withdrawing groups and with varying steric bulk. These reactions yielded either phosphonium salts Ar4P+ or tertiary phosphines Ar3P, with the latter being favoured for more sterically hindered substrates (notably 2-iodotoluene, which provided good conversion to the important ligand (o-tol)3P with good conversion and excellent selectivity; o-tol=2-methylphenyl; Table 2, entry 2),5 as well as electron-deficient substrates (such as methyl 4-iodobenzoate; Table 2, entry 7). Notably, the particularly bulky substrate MesI (Mes=2,4,6-trimethylphenyl) failed to reach even the tertiary phosphine, instead producing the primary and secondary phosphines MesPH2 and Mes2PH as the only products detected by standard 31P{1H} NMR spectroscopic measurements (Table 2, entry 15). The heteroatomic substrate Ph3SnCl could also successfully be employed, providing reasonable, selective conversion to the interesting “P3−” synthon (Ph3Sn)3P without any further reaction optimisation (Table 2, entry 16). In all cases the observed trends in reactivity and selectivity are in excellent agreement with those noted previously for the photocatalytic arylation of P4.9
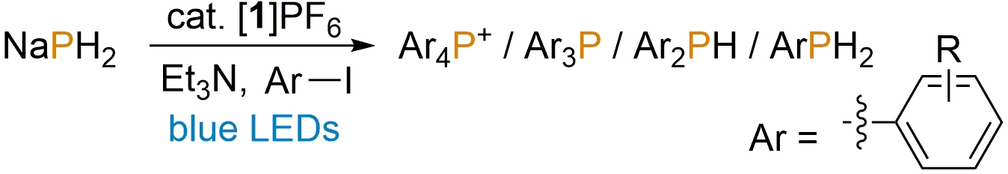
Entry[a] |
R |
Conversion to Ar4P+ [%] |
Conversion to Ar3P [%] |
Conversion to Ar2PH [%] |
Conversion to ArPH2 [%] |
|---|---|---|---|---|---|
1 |
H |
77 |
– |
– |
– |
2 |
2-Me |
– |
63 |
– |
– |
3 |
3-Me |
61 |
6 |
– |
– |
4 |
4-Me |
64 |
<5 |
– |
– |
5 |
2-CO2Me |
– |
10 |
– |
– |
6 |
3-CO2Me |
29 |
<5 |
– |
– |
7 |
4-CO2Me |
– |
32 |
– |
– |
8 |
2-SMe |
– |
13 |
– |
– |
9 |
2-OMe |
– |
42 |
– |
– |
10 |
3-OMe |
53 |
<5 |
– |
– |
11 |
4-OMe |
39 |
6 |
– |
– |
12 |
2-CF3 |
– |
– |
– |
– |
13 |
3-CF3 |
12 |
7 |
– |
– |
14 |
4-CF3 |
7 |
19 |
– |
– |
15 |
2,4,6-Me3 |
– |
– |
11 |
17 |
16 |
Ph3SnCl[b] |
– |
55[c] |
– |
– |
- [a] Reactions performed using 0.1 mmol NaPH2, 2 mol % [1]PF6, 11 equiv ArI, 15 equiv Et3N in 2.0 mL MeCN under blue LED irradiation and N2 atmosphere for 24 h. [b] Ph3SnCl instead of ArI. [c] (Ph3Sn)3P formed instead of Ar3P.
Having established the scope of the [1]PF6-catalysed reaction, the use of the less expensive and more abundantly available, sustainable organic photoredox catalyst 2 was also investigated. Gratifyingly, after only minor further reaction optimisation (Table S4, SI), good conversions could also be achieved using this more practical, precious metal-free catalyst, under similar conditions (Table 3). In general, both catalysts achieve similar conversions, and the same trends in product selectivity are observed with respect to the substrates’ steric and electronic properties. Nevertheless, in several cases an appreciably superior preference is observed for either the metallo- or organo-catalyst (for example: compare entries 2 and 9 in both Table 2 and Table 3), indicating that the choice of catalyst can be tailored to the specific target product.
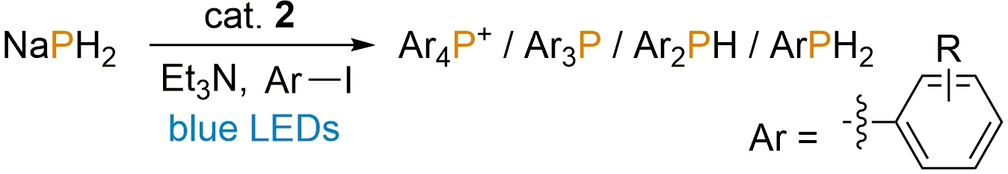
Entry[a] |
R |
Conversion to Ar4P+ [%] |
Conversion to Ar3P [%] |
Conversion to Ar2PH [%] |
Conversion to ArPH2 [%] |
|---|---|---|---|---|---|
1 |
H |
72 |
– |
– |
– |
2 |
2-Me |
– |
51 |
– |
– |
3 |
3-Me |
51 |
<5 |
– |
– |
4 |
4-Me |
56 |
– |
– |
– |
5 |
2-CO2Me |
– |
7 |
<5 |
– |
6 |
3-CO2Me |
20 |
<5 |
– |
– |
7 |
4-CO2Me |
– |
9 |
– |
– |
8 |
2-SMe |
– |
29 |
– |
– |
9 |
2-OMe |
– |
52 |
– |
– |
10 |
3-OMe |
72 |
7 |
– |
– |
11 |
4-OMe |
34 |
14 |
– |
– |
12 |
2-CF3 |
– |
– |
– |
– |
13 |
3-CF3 |
30 |
6 |
– |
– |
14 |
4-CF3 |
<5 |
18 |
– |
– |
15 |
2,4,6-Me3 |
– |
– |
7 |
19 |
16 |
Ph3SnCl[b] |
– |
41[c] |
– |
– |
- [a] Reactions performed using 0.1 mmol NaPH2, 10 mol % 2, 13 equiv ArI, 16 equiv Et3N in 2.0 mL MeCN under blue LED irradiation and N2 atmosphere for 24 h. [b] Ph3SnCl used instead of ArI. [c] (Ph3Sn)3P formed instead of Ar3P.
Mechanisms of photocatalytic arylation
Based on our prior studies of photocatalytic P4, PhPH2 and Ph2PH arylation, as well as the studies described herein, it is possible to propose complete mechanistic courses for both the photocatalytic formation of PH3 from P4 and Et3N, and the photocatalytic arylation of PH3 (Scheme 2). For the former, the photoexcited state of the photocatalyst PC (PC=[1]+ or 2) undergoes reductive quenching by Et3N (confirmed previously by fluorescence quenching experiments) to generate Et3N+. and the reduced catalyst PC.−. Thus generated, PC.− can effect single electron reduction of one of the P-P bonds present in P4 (confirmed previously by UV/Vis spectroelectrochemistry) to generate a phosphorus-centred radical and anion. Abstraction of H. and H+ respectively by these species from Et3N+. (Et3N having been established as the ultimate H atom source for this chemistry; vide supra; for the corresponding in situ NMR study applying fully deuterated Et3N see Section 3.6 in the Supporting Information) then generates new P−H bonds and ultimately PH3.

Complete proposed mechanisms for the photocatalytic reduction of P4 to PH3 in the absence of ArI (a); and the photocatalytic arylation of PH3 in the presence of ArI (b). Here, [P]-[P] indicates a generic P-P bond derived from P4 and [P]-H indicates a generic P−H bond derived from PH3. PC=[1]+ or 2.
Arylation of PH3 is proposed to occur via an analogous mechanism to that already established for P-H arylation of the “downstream” intermediates PhPH2 and Ph2PH. Reduction of ArI by PC.− (generated as outlined above) generates aryl radicals Ar. that can abstract H. to generate transient .PH2 which can then combine with a second equivalent of Ar. to produce ArPH2 (most likely via intermediate formation of the dimer P2H4; c.f. our previous observation of Ph4P2 during P-H functionalisation of Ph2PH).9b Analogous, subsequent arylation steps then generate in turn Ar2PH and Ar3P (and, for certain Ar, Ar4P+), as established in our previous investigations. Formation of this sequence of intermediates has been further confirmed by NMR monitoring of the “single chamber” arylation of NaPH2 (Figure 6).

Reaction profile showing the evolution of P atom speciation during the light-driven phenylation of NaPH2 (0.1 mmol scale) catalysed by organocatalyst 2, as assessed by time-resolved 31P{1H} NMR spectroscopy. Equivalents of reagent (equiv) and catalyst loadings (in mol %) are defined per P atom. Σ(unprod.) indicates the sum of intensities for unproductive side-products Side1–Side5. In the magnified region, the course of PH2− is highlighted as a thicker line to emphasize the very low amounts present throughout.
Notably, the reaction profile in Figure 6 shows that only small amounts of NaPH2 are dissolved at any specific time (PH2−), and also that the sequential reaction steps leading to PPh4+ proceed very cleanly with nearly no side product formation (c.f. Figure 2 for analogous, less selective reactions starting from P4). Based on this apparent correlation it was speculated that using lower concentrations of other monophosphorus starting materials might also provide cleaner reactions, and thus higher eventual yields. And indeed, NMR investigations of the light-driven arylation of H2PPh (applied in different concentrations) confirmed this hypothesis by showing that the total yield of side products increases with increasing concentration of starting material, alongside a concomitant decrease in yield of the final product (for a more comprehensive discussion of side product formation during the arylation of PhPH2, see Section 3.9.4 in the Supporting Information).
Competing mechanisms of P4 arylation
Taken together, the elementary steps outlined above (reduction of P4 to PH3 and its subsequent arylation) provide a plausible alternative mechanistic proposal for our previously reported catalytic arylation of P4. Nevertheless, the mechanistic complexity of these reactions should again be acknowledged, and several specific observations suggest that these are unlikely to be the only kinetically viable elementary mechanistic steps. For example, it was noted in our original report that upon irradiation in the presence of [1]PF6 and Et3N, P4 is consumed more quickly in the presence of PhI than in its absence, with the reverse being observed for consumption of PhI in the presence/absence of P4. While the latter trend is easily explained based upon the above mechanism (since PhI must compete with P4 to react with PC.−), the former is more difficult to account for. Moreover, attempts to quantify the PH3 formed during the photocatalytic reaction between P4 and Et3N in the absence of ArI suggest this to be a rather inefficient process that produces PH3 in only very low yield (see Section 3.12 of the Supporting Information for full details). It is therefore likely that reaction with Ph. can provide an alternative means by which the P-P bonds of P4 are broken (Scheme 3), in line with our initial mechanistic hypothesis.24 We thus propose that initial degradation of the P4 tetrahedron to P1 species can proceed through a combination of both pathways, to generate a mixture of PH3 and ArPH2.25 The relative rates, and hence importance, of these competing mechanistic routes is likely to depend strongly on the identity of the arene substrate (for example, more hindered aryl radicals may add more slowly to P4, and so render this pathway less competitive).

Proposed alternative initial mechanism for the photocatalytic transformation of P4 in the presence of ArI. Here, [P]-[P] indicates a generic P-P bond derived from P4.
Conclusion
Detailed NMR spectroscopic studies have been used to provide a significantly deeper and more comprehensive understanding of the mechanism of our recently reported, unprecedented direct photocatalytic arylation of P4. Despite the complexity of this elaborate transformation, it is now possible to propose a plausible and comprehensive set of elementary steps for this reaction, supported by experimental observations. In addition to these primary productive steps, it has been shown that a wide variety of competing side reactions are relevant to the overall course of the reaction, resulting in of a large number of minor side products and intermediates. While these products are present in individually insignificant quantities they sum to an appreciable fraction of the overall P-containing reaction products, and their formation therefore explains the inability of these transformations to provide quantitative yields of their target products, despite full consumption of starting material and apparently clean conversion. Moreover, the use of the organophotocatalyst 2 to mediate these reactions instead of the iridium catalyst [1]PF6 has been shown to significantly increase the prevalence of these side products which provides an explanation for its poorer catalytic performance in P4 arylation, despite other investigations showing it to be a superior catalyst for the closely related arylation of PhPH2 and Ph2PH. These side products seem to be primarily derived from unexpected side reactions of the Et3N reductant (or the aminium, iminium or enamine oxidation products thereof). Given the widespread use of Et3N as an electron donor in photoredox chemistry, these results provide further evidence that similar side reactions may have an underappreciated limiting effect on reaction yields for many other transformations. Based on this realisation, current efforts are being made to replace Et3N with a “better-behaved” combination of reductant and hydrogen source, to facilitate the desired transformation while minimising undesired side reactivity.
Alongside this mechanistic insight, based on the detection of PH3 as an additional intermediate, these studies have also enabled the development of an entirely new photocatalytic transformation. Specifically, the first examples of the catalytic arylation of PH3 have been developed, which allow for the unprecedented direct transformation of this key industrial P1 precursor into a sterically and electronically diverse range of valuable triarylphosphines and tetraarylphosphonium salts. Notably, this reaction does not require a precious metal catalyst, and works well using 3DPAFIPN as an inexpensive and more sustainable organic photoredox catalyst.
Acknowledgements
We thank Gabor Balász (University of Regensburg) for a generous donation of NaPH2 and Burkhard Luy (Karlsruhe Institute of Technology) for providing the broadband pulse xyBEBOP. The project was funded by the European Research Council (ERC CoG 772299), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR 325 – 444632635, and the Alexander von Humboldt Foundation (postdoctoral fellowship for D.J.S.). Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.

