

Oxidative Addition of Water, Alcohols, and Amines in Palladium Catalysis
Graphical Abstract
The oxidative addition of OH, NH, and weak CH bonds to a palladium(0) complex is facile, allows for additive-free catalysis, and suggests the non-innocence of water in palladium catalysis. The oxidative addition of protic solvents or adventitious water switches the chemoselectivity in catalysis with alkynes through activation of the terminal C−H bond.

Abstract
The homolytic cleavage of O−H and N−H or weak C−H bonds is a key elementary step in redox catalysis, but is thought to be unfeasible for palladium. In stark contrast, reported here is the room temperature and reversible oxidative addition of water, isopropanol, hexafluoroisopropanol, phenol, and aniline to a palladium(0) complex with a cyclic (alkyl)(amino)carbene (CAAC) and a labile pyridino ligand, as is also the case in popular N-heterocyclic carbene (NHC) palladium(II) precatalysts. The oxidative addition of protic solvents or adventitious water switches the chemoselectivity in catalysis with alkynes through activation of the terminal C−H bond. Most salient, the homolytic activation of alcohols and amines allows atom-efficient, additive-free cross-coupling and transfer hydrogenation under mild reaction conditions with usually unreactive, yet desirable reagents, including esters and bis(pinacolato)diboron.
Introduction
The homolytic activation of a bond through oxidative addition is an important chemical transformation. It is in particular central to organometallic chemistry, where the insertion of low-valent, late transition metal complexes into, most typically, carbon-halogen bonds has revolutionized homogeneous catalysis.1 The catalytic valorization of O−H, N−H and C−H bonds is equally important in catalysis. For instance, the dehydrogenation of alcohols or the hydrogen generation from water hold promise to cleanly convert and store energy.2 In the vast field of molecular palladium catalysis, it is however common belief that O−H, N−H and weak C−H bonds are only cleaved heterolytically through deprotonation with a base. Strong indication for this redox-neutral mechanism was presented by μ-hydroxo bridged palladium(II) dimers in Wacker-type oxidation reactions and the Buchwald–Hartwig amination.3 These very basic palladium(II) hydroxo complexes deprotonate anilines to give water and the μ-anilido bridged palladium(II) dimers (Scheme 1 A).4

Activation of N−H or O−H bonds by either deprotonation (A) or oxidative addition (B).
The alternative oxidative addition, in case of water referred to as water splitting,5 produces a palladium(II) hydride from a palladium(0) precursor (Scheme 1 B). This palladium(II) hydride is the alleged key intermediate for subsequent redox transformations including the generation of dihydrogen or dehydrogenative cross coupling.6 Nevertheless, whereas the intermolecular oxidative addition of strong O−H and N−H bonds is known for the group 9 metals,7 it remains elusive for the group 10. To the best of our knowledge, the only exceptions are the 1,2-addition across activated, nucleophilic alkylidene complexes,8 a transient palladium(I) complex with a pincer ligand,9 and the solvolysis of bis(triisopropylphosphine) platinum(0).10 Eventually, it is intriguing to note that protic solvents or co-reagents sometimes play a pivotal role in palladium catalysis. For example, aqueous reaction media can lead to exceptional efficient and robust cross-coupling;11 similarly, hexafluoroisopropanol (HFIP) emerged as a favorable solvent in C−H functionalization chemistry.12 Nevertheless, the oxidative addition of adventitious water or alcohols in general seems to not have been considered in this context. In fact, water or alcohols have been reported to instead mediate the reductive generation of palladium(0) from palladium(II) precursors13 with phosphane- or N-heterocyclic carbene (NHC)14 ligands.
Results and Discussion
We recently used a palladium(0) complex with an ancillary cyclic (alkyl)(amino)carbene (CAAC) ligand15 as a reactive intermediate.16 The solid state structure reveals now that 1 is a mononuclear, pyridine-coordinate 14-electron complex without interaction of the metal with the pendant imino group (Figure 1, left).

Complex 1 parallels the alleged palladium(0) intermediate of the PEPPSITM precatalyst family.41 Ellipsoids are shown at the 50 % probability level, a second independent molecule of 1 and hydrogen atoms are omitted for clarity. Selected bond lengths [Å], angles [°] and dihedral angles between least-square planes [°]: Pd1–C1 1.927(3), Pd1–N3 2.092(3); C1-Pd1-N3 174.6(2), N1-C1-N3-C41 81.1. The second independent molecule shows a dihedral angle of 47.3° between least-square planes of N4-C42-N6-C82.
Two independent molecules of 1 with different dihedral angles between least-squares planes, which were calculated through the five atoms forming the CAAC and the six atoms of the pyridino ligand (81.1° and 47.3°), were found in the unit cell, thus indicating at best weak π-backbonding from the metal to the pyridino ligand. Pyridine-coordinated linear palladium(0) complexes have previously not been reported to the best of our knowledge. In fact, dicoordinated, heteroleptic palladium(0) complexes are very rare and only examples for soft phosphine ligands in combination with likewise soft NHCs are known.17 Complex 1 represents thus an excellent model for the alleged palladium(0) intermediate of the PEPPSITM palladium(II) NHC precatalyst family (Figure 1, right), which show outstanding activity in a variety of important catalytic transformations.18 Most intriguingly, we reasoned that the soft/hard mismatch between the low-valent metal and the pyridine ligand renders 1 a surrogate for the elusive 12-electron palladium(0) intermediate in homogeneous catalysis.19
Truly, the very broad 1H NMR signals of 1 in perdeuterated benzene suggest dissociation of the pyridine and potential formation of aggregates. Stirring solutions of 1 in benzene at room temperature in the presence of 15 equivalents of water led to a color change from dark red to yellow, which is indicative for the oxidation to a palladium(II) complex. The conversion of 1 was monitored by 1H NMR spectroscopic analysis and a singlet at −5.16 ppm suggested the quantitative formation of a palladium hydroxo complex. Indeed, the solid-state structure revealed the formation of the bis-μ-hydroxo bridged dimer dimer2H2O (Figure 2).

Solid-state structure of dimer2H2O.41 Ellipsoids are shown at the 50 % probability level, solvent molecules and hydrogen atoms are omitted for clarity (except the ones connected to the carbene C atom and the OH group). Selected bond lengths [Å], angles [°] and dihedral angles [°]: Pd1–Pd2 3.1902(2), Pd1–O1 2.041(2), Pd1–O2 2.195(2), Pd2–O2 2.045(2), Pd2–O1 2.183(2), Pd1–C1 1.989(2), Pd2–C37 1.995(2), Pd1–N2 2.029(2), Pd2–N4 2.027(2); N2-Pd1-C1 82.11(8), N4-Pd2-C37 82.51(8), O1-Pd1-O2 75.08(6), Pd1-O1-Pd2, 98.05(6), O1-Pd1-C1-N1 71.2(2), O2-Pd2-C37-N3 69.4(2).
The formation of a bond between the highly electrophilic pz orbital of the CAAC ligand and the hydrogen atom derived from water is intriguing. It suggests, analogous to the hydride migration observed upon coordination of free CAACs to aluminum- or borohydride,20 the previous formation of the palladium (hydrido)(hydroxo) intermediate 2(H)(OH) (Scheme 2). The equilibrium of the overall reaction turned out to be solvent dependent. Dissolving dimer2H2O in pyridine resulted in the clean regeneration of 1 with the concomitant release of one equivalent of water within six days (see Figure S1 in the Supporting Information).

The equilibrium between 1 and 2H2O is solvent dependent.
Further testing the reversibility of the reaction, we treated dimer2H2O in tetrahydrofuran with 80 equivalents of deuterated water (Figure S2). Following instantaneous isotopic exchange of the protons of the bridging hydroxo ligands, we also observed the isotopic exchange of the hydrogen atom connected with the former carbene atom over the course of one week (60 °C: 3 h).
Complex dimer2H2O was treated with hydrogen chloride in order to unambiguously validate its hydridic reactivity (Scheme 3). Upon addition of one equivalent of hydrogen chloride, the exchange of the hydroxo- for a chloro-ligand and the formation of dimer2HCl was observed. Subsequent addition of another equivalent of hydrogen chloride led to the immediate formation of gas bubbles, which were identified as dihydrogen, as well as the precipitation of 3, which was isolated in quantitative yield (Figure S5).

Complexes 2ROH show the reactivity of palladium hydrides.
For the analogous phenolate complex 2PhOH, stoichiometric amounts of phenol, dihydrogen and 3 were obtained (see Figure S6). Starting from deuterated dimer2D2O gave quantitatively hydrogen deuteride HD (Figures S7 and S8). Hydrogenated CAACs embedded in extended π-systems require strong oxidants to be dehydrogenated and the addition of acids does not lead to dihydrogen generation.21 Equally note that α-hydride elimination is an established strategy to synthesize Fischer-carbene complexes, including aza-Fischer carbenes isostructural with CAACs.22 Overall, we conclude that dimer2H2O shows the reactivity of a palladium hydride and is likely to stand in equilibrium with 2(H)(OH).
In order to further elucidate the water activation mechanism, we determined a kinetic isotope effect k of 2.0 (Figures S9 and S10). This value is consistent with the value of k
of 2.0 (Figures S9 and S10). This value is consistent with the value of k =2.0 found for the oxidative addition of methanol to an iridium(I) complex.7a Varying the concentration of water revealed that the reaction follows a second order rate law in regard to water in tetrahydrofuran (Figure 3, top left). This is also in agreement with the mechanism found for the iridium(I) complex, where a reaction order “higher than one” was reported.7a The conversion of 1 in the presence of 100 equivalents of water initially follows pseudo-first-order kinetics, but then shows product inhibition (Figure 3, bottom; linearization according to Lineweaver—Burk: top right). This indicates a slower reaction rate due to the liberation of pyridine (cf. Table 1) and hence dissociation of pyridine prior to the rate determining transition state.
=2.0 found for the oxidative addition of methanol to an iridium(I) complex.7a Varying the concentration of water revealed that the reaction follows a second order rate law in regard to water in tetrahydrofuran (Figure 3, top left). This is also in agreement with the mechanism found for the iridium(I) complex, where a reaction order “higher than one” was reported.7a The conversion of 1 in the presence of 100 equivalents of water initially follows pseudo-first-order kinetics, but then shows product inhibition (Figure 3, bottom; linearization according to Lineweaver—Burk: top right). This indicates a slower reaction rate due to the liberation of pyridine (cf. Table 1) and hence dissociation of pyridine prior to the rate determining transition state.

Top left: The activation of water follows a second-order rate law in respect to water. Bottom: The formation of dimer2H2O (48 mmol L−1 1 in [D8]THF; 100 equiv. H2O; in-situ 1H NMR) follows initially a pseudo-first-order rate law followed by product inhibition. Top right: Linearization according to Lineweaver—Burk (see Figure S61 for further details).
Reagent |
Equiv. Reagent[a] |
Solvent |
Pseudo-first-order rate constant kobs [s−1] |
|---|---|---|---|
H2O |
15 |
C5D5N |
no reaction |
H2O |
15 |
[D8]THF |
(8±2)×10−6 |
H2O + 15 equiv. C5H5N |
15 |
[D8]THF |
(5±1)×10−7 |
H2O[b] |
15 |
C6D6 |
(6±5)×10−5 |
H2O |
2 |
[D8]THF |
(9±3)×10−8 |
H2O[c] |
2 |
C6D6 |
(2±2)×10−6 |
iPrOH[d] |
15 |
[D8]THF |
(9±4)×10−7 |
PhNH2[d] |
15 |
[D8]THF |
(5±3)×10−5 |
PhOH |
2 |
C5D5N |
(6±4)×10−4 |
HFIP |
2 |
C5D5N |
(2±1)×10−3 |
PhCl |
2 |
C5D5N |
(9±3)×10−4 |
HCl |
1 |
C6D6 |
>1×10−2 |
ArCCH[e] |
1 |
C5D5N |
π-complex |
- [a] Relative to 1. [b] Emulsion.24 [c] Homogeneous. [d] Reaction gives a mixture of products in C6D6 and [D8]THF due to reversible ligand redistribution; the product was isolated from a reaction in neat isopropanol or aniline, respectively; see Figures S4, S11, and S12 for further details. [e] Ar=4-tert-butylphenyl-.
The same dissociative substitution mechanism via a formal 12-electron palladium(0) intermediate has been suggested for the oxidative addition of aryl halides to palladium(0) bisphosphine complexes.23
The reaction rate for the oxidative addition of water also showed a strong dependence on the solvent (Table 1).While no reaction was observed in pyridine, an initial pseudo first order rate constant of kobs=(8±2)×10−6 s−1 (15 equiv. of water) was determined in tetrahydrofuran. Adding 15 equiv. of pyridine (Table 1) decreased the reaction rate (kobs=(5±1)×10−7 s−1). In benzene, a faster reaction was obtained, both with 15 equiv. of water, which is an emulsion, or with 2 equiv. of water, which is a homogeneous reaction, (15 equiv. in benzene: kobs=(6±5)×10−5 s−1, 2 equiv. in benzene: kobs=(2±2)×10−6 s−1, 2 equiv. THF: kobs=(9±3)×10−8 s−1). We subsequently evaluated the reaction rates with isopropanol, aniline, phenol, HFIP and hydrogen chloride and compared them with the common oxidative addition of chlorobenzene. Following the reduced acidity of the protic reagents, isopropanol (15 equiv. in THF: kobs=(9±4)×10−7 s−1) and aniline (15 equiv. in THF: kobs=(5±3)×10−5 s−1) reacted with a slower rate than phenol (2 equiv. in pyridine: kobs=(6±4)×10−4 s−1). The oxidative addition of chlorobenzene (2 equiv. PhCl in pyridine: kobs=(9±3)×10−4 s−1) proceeded with the same order of magnitude as was found for phenol (2 equiv. phenol in pyridine: (6±4)×10−4) and HFIP (2 equiv. HFIP in pyridine: kobs=(2±1)×10−3 s−1). We hence conclude that O−H oxidative addition of HFIP seems to be a likely reaction upon generation of palladium(0) in this solvent. The reaction with hydrogen chloride went to completion before we could record an NMR spectrum (kobs>1×10−2 s−1), which is consistent with previous reports in the literature.25
Complex 2PhOH shows a monomeric structure in solution, while 2HCl forms dimers like dimer2H2O according to diffusion NMR measurements (Table S1). 2PhOH is also a monomer in the solid state with a rare tricoordinate, T-shaped coordination geometry and hence a vacant coordination site (Figure 4).

Solid-state structure of 2PhOH.41 Ellipsoids are shown at the 50 % probability level, solvent molecules and hydrogen atoms (except the one connected to C1 and the hydroxy group) are omitted for clarity. Selected bond lengths [Å], angles [°] and dihedral angles [°]: Pd1–C1 2.026(2), Pd1–O1 2.0586(8); N2-Pd1-C1 83.28(4), N2-Pd1-O1 177.08(4), O1-Pd1-C1-N1 66.69(6).
Intriguingly, following the formation of 2PhOH in deuterated pyridine by 1H NMR spectroscopic analysis (Figure 5, Figures S13, S14) revealed a signal of low intensity at −14.93 ppm (≈20 % relative to 2PhOH at 10 % conversion). This signal, which is in the common range for palladium hydrides,26 disappeared over the course of the reaction. An analogous signal (−13.72 ppm in [D8]THF) was observed in the reaction with aniline (see Figure S15), but not with water. We suggest that these signals relate to the fleeting intermediates 2(H)(OPh) and 2(H)(HNPh), respectively (vide infra).26 Eventually, despite the comparably high acidity of terminal acetylenes, the reaction with 4-tert-butylphenylacetylene did not give the oxidative addition product, but instead the π-complex 4 (Scheme 4, left). However, the reaction of dimer2H2O with 4-tert-butylphenylacetylene (Scheme 4, right) afforded cleanly 2HCCAr in analogy to Hartwig's deprotonation of anilines (cf. Scheme 1).4 This reaction sequence reveals that water mediates the formal oxidative addition of a weak C−H bond.

A palladium hydrido complex is the intermediate for the formation of 2PhOH from 1. Integration is given in relation to reaction product 2PhOH. Blue, prior to addition of phenol; green, 10 % conversion; red, 35 % conversion, pink, 55 % conversion; yellow, 100 % conversion. See Figures S13–S15 for the entire spectra and the reaction with aniline.

Water catalyzes the oxidative addition of 4-tert-butylphenylacetylene. Solid-state structures of 4 and 2HCCAr.41 Ellipsoids are shown at the 50 % probability level, solvent molecules and hydrogen atoms are omitted for clarity, except for 4: connected to C37 and 2HCCAr: connected to C1. Selected bond lengths [Å], angles [°] and dihedral angles [°] 4: Pd1–N2 2.258(4), Pd1–C1 1.987(4), Pd1–C38 2.050(4), Pd1–C37 1.999(4); C1-Pd1-N2 76.0(2), C38-Pd1-C37 36.7(2); 2HCCAr Pd1–C1 2.018(2), Pd1–N2 2.050(2), Pd1–C37 1.987(2); C1-Pd1-N2 82.63(7), C37-Pd1-C1 98.17(8), C37-Pd1-N2 172.63(8).
We modeled the reactions in silico in order to further elucidate the mechanism (Scheme 5, black).27 The first elementary step is the substitution of the pyridino- by an aqua ligand (ΔG=+29 kJ mol−1; ΔG≠=+62 kJ mol−1). Subsequent oxidative addition to 1H2O involving two molecules of water (ΔG≠=+108 kJ mol−1) is the rate determining transition state. The (hydrido)(hydroxo) complex 2(H)(OH) (ΔG= +20 kJ mol−1) was predicted to be kinetically unstable and to give 2H2O (ΔG=−6 kJ mol−1; ΔG≠=+99 kJ mol−1) via migration of the hydride to the carbene.

Most favorable28 reaction profile for the activation of water (black) and phenol (purple) in tetrahydrofuran (PBE0-D3BJ(THF)/def2-TZVPP//PBE0-D3BJ/def2-SVP). For aniline, other solvents (pyridine, benzene), and further details, see Figures S62–71 and Tables S9–12.
Numerous further reaction pathways (Scheme 5, Figures S67 and S68), including oxidative addition involving only one molecule of water (ΔG≠=+121 kJ mol−1), an assisting molecule of pyridine (ΔG≠=+114 kJ mol−1) and two molecules of water in a 1,2-addition mechanism across the carbene metal bond (ΔG≠=+131 kJ mol−1) are less favorable. Eventually, note that the dimerization 2H2O further shifts the equilibrium towards the product side (Figure S69). The calculations confirm (Figure S70) that the reaction proceeds with a lower barrier in benzene (ΔG≠= +106 kJ mol−1; ΔG=−3 kJ mol−1) than in tetrahydrofuran (ΔG≠=+108 kJ mol−1; ΔG=−6 kJ mol−1, vide supra). Contrarily, it is predicted to proceed endergonic (ΔG≠= +25 kJ mol−1) in pyridine with a very high activation energy (ΔG≠=+139 kJ mol−1). The computational results for the oxidative addition of phenol (Scheme 5, purple) are likewise in excellent agreement with the experiment. They corroborate (I.) a very fast reaction at room temperature and (II.) the spectroscopic capture of the fleeting (hydrido)(phenolato) intermediate 2(H)(OPh) (ΔG=−16 kJ mol−1; ΔG≠= +89 kJ mol−1 and +58 kJ mol−1, respectively). In further agreement with the experiment a fairly similar barrier of ΔG≠= +70 kJ mol−1 is calculated for the oxidative addition of chlorobenzene (ΔG=−127 kJ mol−1; Figure S77).
A closer look at the structural parameters of the rate determining transition state ts1H2O-2(H)(OH) suggests that the hemilabile imino ligand29 provides only moderate stabilization to the transition state (Pd−N: 2.46 Å; Figure 6) and rather “traps” the final oxidative addition product through coordination to give a square planar palladium(II) complex.30 This suggests that the oxidative addition of water should proceed with slightly slower kinetics for “conventional” NHCs and CAACs. Hence and in order to quantify the effect of the hemilabile imino group on the kinetics, the related transition states for the NHC with two diisopropylphenyl substituents (IPr) as well as a CAAC with two methyl groups adjacent to the carbene atom (CAACMe) were calculated. The reaction barrier for the water activation in tetrahydrofuran was predicted to be ΔG≠=+128 kJ mol−1 (ΔG≠= +121 kJ mol−1 in benzene) for the NHC and ΔG≠=+126 kJ mol−1 (ΔG≠=+119 kJ mol−1 in benzene) for the CAAC complex (see Figures S72, S73). These values suggest that the activation of water by the PEPPSITM system might require gentle heating or a higher water concentration at room temperature.

Structural parameters of the rate-determining transition-state ts1H2O-2(H)(OH). Calculated bond lengths are given in [Å].
Eventually, we validated the mechanism for the formation of dihydrogen after the addition of acids, that is, the protonation of dimer2HCl (Scheme 6). The calculations confirm that direct dihydrogen generation from 2(HCl)+ is not feasible (ΔG≠=+139 kJ mol−1). Contrarily, the α-hydride elimination to give 2(H)(HCl)+ is very facile (ΔG≠= +84 kJ mol−1, ΔG=+2 kJ mol−1) even at room temperature. The subsequent dihydrogen release proceeds essentially barrierless (ΔG≠=+7 kJ mol−1, ΔG=−44 kJ mol−1). We again conclude that the intermediate palladium hydride is key for the dihydrogen generation.

Dihydrogen evolution with acids proceeds in tetrahydrofuran via a palladium hydride (PBE0-D3BJ(THF)/def2-TZVPP//PBE0-D3BJ/def2-SVP). See Figures S74 and S75 for less favorable isomers of 3+ and transition states.
Encouraged by the facile and reversible oxidative addition of various reagents to 1, we evaluated a series of desirable catalytic transformations: In order to demonstrate reversible hydride transfer as required for reduction chemistry and hydrogen storage, we studied the transfer hydrogenation of acetophenone in isopropanol (Scheme 7). This reaction, which typically requires the addition of co-reagents such as a base, is very well established for the group 9 metals. It proceeds there via metal hydrides, but examples for group 10 elements are scarce.31 Indeed, complex 1 readily catalyzed the reduction of acetophenone under mild reaction conditions (0.5 mol % 1, 65 °C, 6 h: 75 %; 22 h: 90 % conversion) without the addition of further co-reagents, whereas no conversion was obtained in blind reactions (a) without complex 1, (b) with only the ligand or (c) in presence of 5 mol % NaOH. Thereby, the 1H NMR spectroscopic analysis confirmed that 2iPrOH is the resting state of the catalyst (Figure S36).

Catalytic transfer hydrogenation of acetophenone in isopropanol. For further details see Table S2 and Figures S35–37.
Amides are typically synthesized from activated carboxylic acid derivatives or in the presence of an over-stoichiometric amount of a strong base.32 The cross coupling of esters with amines is challenging and was only realized for palladium and nickel NHC complexes under harsh reaction conditions (Pd: 3 mol % cat., 1.5 equiv. K2CO3, 110 °C, 16 h; Pd-PEPPSI33 and [Pd(IPr)(acac)Cl]34 similar conditions; Ni: 15 mol % cat., 1.25 equiv. Al(OtBu)3, 60 °C, 12 h).35 Just recently, a protocol using milder conditions (Pd: 1 mol % cat., 2 equiv. Cs2CO3, 40 °C, 4 h) was found.36 We hypothesized that the facile oxidative addition of the N−H bond of aniline should allow for the amidation of an ester under mild conditions even without additives. Indeed, we observed the formation of N-phenylacetamide 5 in 85 % crude yield after 48 h reaction time at only 66 °C (1 h: 33 %; 24 h: 80 %; Table 2).
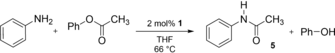
t [h] |
Modification |
Yield [%][a] |
|---|---|---|
1 |
– |
33 |
24 |
— |
80 |
48 |
- |
85 (84)[b] |
24 + 24 |
Addition of 0.3 equiv. KOtBu after 24 h |
97 (96)[b] |
24 h |
absence of 1 |
0 |
- [a] Yield determined by in situ 1H NMR spectroscopy. [b] Yield of isolated product.
No conversion was observed in the absence of 1. Of course, this reaction also showed product inhibition due to the liberation of phenol and subsequent oxidative addition to 1. Accordingly, the equilibrium of the reaction could be further shifted by addition of 0.3 equiv. of potassium tert-butoxide (KOtBu), that is, deprotonation of 0.3 equiv. of phenol (97 % crude yield). The direct formation of mixed borate esters from alcohols and bis(pinacolato)diboron (B2Pin2) is equally challenging, because it involves the oxidative addition of either a strong O−H or B−B bond.37 However, metal alkoxides react readily with B2Pin2.38 Indeed, 0.5 mol % of 1 allowed for the unprecedented and quantitative conversion of B2Pin2 and phenol, aniline, tert-butanol, tert-butylamine, n-octanol and n-octylamine to dihydrogen and the respective (amino-)borates 6ER. All reactions proceeded at room temperature, with for example 20 % conversion for phenol, within 1 h, and achieved quantitative conversion at 50 °C within less than 2.5 h (Table 3, Figures S40–53). Of course, no turnover was obtained in the absence of 1 or the presence of the ligand only.
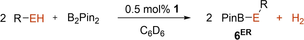
E |
R |
t [h] |
T [°C] |
Yield [%][a] |
|---|---|---|---|---|
O |
Ph |
1.5 |
50 |
>99 |
NH |
Ph |
2.5 |
50 |
>99 |
O |
tBu |
1 |
50 |
>99 |
NH |
tBu |
1 |
50 |
>99 |
O |
n-oct. |
1 |
rt |
>99 |
NH |
n-oct. |
1 |
rt |
>99 |
- [a] Yield determined by in situ 1H NMR spectroscopy.
We eventually explored the catalytic valorization of 4-tert-butylphenylacetylene in order to expose the oxidative addition of adventitious water in palladium catalysis. Palladium(II) catalysts with a vacant coordination site polymerize terminal alkynes,39 whereas Pd0/PdII catalytic cycles were proposed for the dimerization to enynes.40 Indeed, 1 catalyzes the selfhydroalkynylation to dimer 7 in 78 % crude yield (isolated yield: 65 %) under dry conditions (Scheme 8 A; Figures S54–56). Conversely, using the same reaction conditions except for the addition of varying amounts of water (1–100 equivalents) gave quantitatively trans-cisoidal poly(arylacetylene) 8 (Scheme 8 B; Figures S57-58 and Table S7). Catalytic amounts of either dimer2H2O or 2HCCAr afforded the polymer as well. Apparently, and as supported by the computations (Figure S76), the oxidative addition of water shuts down the palladium(0) pathway, mediates the formation of 2HCCAr and consequently switches the chemoselectivity.

A) Catalytic dimerization (head to head, E) of terminal acetylene under dry conditions. B) Catalytic polymerization of terminal acetylene.
Conclusion
We report the facile oxidative addition of N−H and O−H bonds, including water and aniline, to a palladium(0) complex to give reactive palladium(II) hydrides. The bond activation proceeds via a cyclic transition state featuring two substrate molecules and parallels mechanistically an intramolecular deprotonation. These reactions are reversible at room temperature with rate constants similar to the oxidative addition of chlorobenzene. The investigated complex, a surrogate of the elusive 12-electron intermediate in palladium catalysis, resembles the alleged active species of pervasive N-heterocyclic carbene (NHC) palladium(II) precatalysts. The introduction of a hemilabile imino group allows trapping of otherwise transient intermediates and hence suggests that the homolytic activation of strong N−H and O−H bonds might also be of relevance for other palladium catalytic systems with more common ligands. The catalytic transfer hydrogenation of secondary alcohols illustrates additive-free and reversible hydrogen transfer chemistry. Indeed, the complex also catalyzes the challenging direct borylation of alcohols and amines with bis(pinacolato)diboron under release of dihydrogen and the cross coupling of an ester with aniline. All these reactions proceed under mild reaction conditions and without the addition of co-reagents such as a base. Eventually, we elucidate the effect of protic solvents or reagents on the functionalization of a terminal alkyne. We find that water catalyzes the formal oxidative addition of the terminal C−H bond, which then switches the chemoselectivity from hydroalkynylation to alkyne polymerization.
Overall, this work discloses the oxidative addition of O−H and N−H bonds as a synthetic strategy in palladium chemistry, illustrates cyclic (alkyl)(amino)carbene (CAAC) ligand cooperativity with a hydrogen atom, outlines how to achieve efficient catalysis in absence of additives, and describes the non-innocence of adventitious water for the activation of a weak C−H bond.
Acknowledgements
We thank the Fonds der Chemischen Industrie (Liebig fellowship for D.M.) as well as the Bavarian Equal Opportunities Sponsorship—Realization of Equal Opportunities for Women in Research and Teaching (A.G.) for financial support. We thank the RRZ Erlangen for computational resources. Support by K. Meyer is gratefully acknowledged. We also thank Ro. Dorta, S. Harder, and M. Driess as well as S. Schneider for helpful discussions. Open access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.

