

Biocatalysis: Enzymatic Synthesis for Industrial Applications
Graphical Abstract
Biocatalysis has developed into a mature technology for chemical and pharmaceutical synthesis as well as other areas where high selectivity and mild reaction conditions are required. This Review highlights recent achievements with a special focus on industrialized applications including the introduction of key performance indicators (KPIs) to judge the efficiency of enzymes.

Abstract
Biocatalysis has found numerous applications in various fields as an alternative to chemical catalysis. The use of enzymes in organic synthesis, especially to make chiral compounds for pharmaceuticals as well for the flavors and fragrance industry, are the most prominent examples. In addition, biocatalysts are used on a large scale to make specialty and even bulk chemicals. This review intends to give illustrative examples in this field with a special focus on scalable chemical production using enzymes. It also discusses the opportunities and limitations of enzymatic syntheses using distinct examples and provides an outlook on emerging enzyme classes.
1 Introduction
Biocatalysis has developed in the last two decades into a rather mature and widely used technology.1 With a few noticeable exceptions, biocatalysis in the early 2000s kept hiding in niche applications and focused on the synthesis or resolution of optically active intermediates.2 Since then, biocatalysis has evolved more and more into a broadly applicable tool for chemical synthesis and manufacturing as documented in many books.3 Important driving forces are the rapid discovery of new enzyme variants by modern bioinformatics and computer modelling supported enzyme engineering.4 While the tremendous catalytic activity of enzymes is widely recognized, often their stability and cost are considered a limitation. In this review we will focus on biocatalysis suitable for scalable chemical production and discuss the opportunities and limitations of enzymatic syntheses using distinct examples.
A search in Scopus® (Elsevier) for reviews on “Biocatalysis” reveals more than 2000 hits; in SciFinder® (Chemical Abstract Service, CAS) with a less stringent definition of “review” more than 5000 articles are documented. Our aim shall not be to add yet another review simply summarizing the latest achievements in the biocatalysis field. Instead, we rather intend to give guidance to synthetic chemists which biocatalytic conversion technology may serve his/her manufacturing challenge best. For this purpose, important key performance indicators (KPIs) will be applied to provide efficiency considerations that qualify new biocatalytic processes for industrial scale-up and commercialization. While we will give reference to more specialized reviews of the individual biotransformations, our comprehensive approach shall help synthetic chemists navigate to the most efficient route for a multistep synthesis involving biocatalysis.
When in the early 2000s seminal reviews appeared,2 biocatalysis was still mostly using hydrolases (such as lipase CAL-B) or amidases (such as penicillin acylase and Subtilisin), predominantly for the kinetic resolution of chiral primary and secondary alcohols, amines or carboxylic acids. Ketoreductases (KREDs, as a subgroup of alcohol dehydrogenases, ADHs) were employed to make chiral secondary alcohols via asymmetric reduction of prochiral ketones. Probably the most prominent large scale industrial biocatalytic process was the long-established nitrile hydratase (NHase) process to make acrylamide from acrylonitrile.5 Here, the NHase from Rhodococcus rhodochrous J1—used in a whole-cell system to avoid enzyme isolation as no undesired side reactions occur and stability is higher—exhibits outstanding catalytic efficiency as up to 7 kg acrylamide can be produced per gram cells with product concentrations exceeding 500 g per liter reactor volume and space-time-yields (STY) exceeding 0.1 kg L−1 h−1.
In the meantime, many more enzymes made it into large scale biocatalytic processes for which several examples are given in this review. One reason is faster and straight forward discovery and engineering of suitable biocatalysts (the 3rd[1a] and 4th[6] “wave”). This includes access to a plethora of novel enzymes via protein sequence and structure databases, their improvement guided by bioinformatic tools in combination with rational design or directed evolution, high-throughput screening tools as well as a range of design methods as summarized in reviews.7 Especially directed evolution represents a key technology for which Frances H. Arnold was awarded the Nobel prize in Chemistry in 2018.8 The major acceleration of biocatalyst development in recent years stems from the cheap availability of synthetic genes that allow for rapid, affordable screening of a diverse set of enzyme variants. In addition, the strategic planning of enzymatic routes has been facilitated as several reviews9 and a book10 now cover retrosynthesis concepts for biocatalysis, which should ease the decision of which type of enzyme (class) and reaction is most suitable for a targeted product. Also, the combination of biocatalysis with chemical catalysis (metal-, organo-, photo-, electro-catalysis) became more mature in the past decade.3b
Still, not every new biocatalytic reaction (theoretically) possible or working on small scale makes it into an industrial process for various reasons, as also pointed out by Hauer very recently.11 Many of these reasons also apply to new chemical reactions, which never make it into production. For instance, it can be difficult to get a new process implemented simply because this requires new investments into a factory while an old process in a depreciated production site is still running profitably. Furthermore, despite the achievements made in enzyme discovery and engineering, the “need for speed” can still be an issue, as timelines for biocatalyst development especially in the pharmaceutical industry are often very short as stated in an excellent recent publication.12 Other aspects are given in Table 1. On the other hand, biocatalytic reactions have the advantage that no special equipment is required and reactors commonly applied for chemical synthesis can be used.
Problem statements |
Solutions |
|---|---|
Enzymes are expensive, not all are available |
Recombinant expression in a suitable (microbial) host, either in-house or with specialized enzyme producer company, immobilization to facilitate re-use and cost reduction |
|
|
Enzymes are unstable |
Enzyme engineering via rational design or directed evolution, immobilization to enhance stability |
|
|
Dependency on expensive cofactors |
For NADH, NADPH and more recently also for ATP, efficient recycling systems are available and demonstrated on industrial scale |
|
|
Development time is too long |
Use interdisciplinary teams for planning of best chemical route for integrated enzyme engineering and process development early enough |
|
|
Process development and down-stream processing are difficult |
Numerous examples and concepts for bioreaction engineering are available |
An interesting example where an initially considered “expensive” enzymatic process has replaced a “simple and cheap” chemical reaction has been developed by the company Goldschmidt (now Evonik) is in the synthesis of emollient esters (e.g., myristyl myristate or coconut oil esters): despite the use of a rather expensive enzyme (immobilized lipase CAL-B, which is recycled multiple times to reduce costs), especially consideration of the entire process and not only the ester synthesis step made the enzymatic route cost-efficient. The chemical process took place at high temperatures (>180 °C), which beside higher energy costs compared to the lipase-catalyzed process (60–80 °C) caused formation of (smelly and coloured) by-products. This required several downstream processing steps such as deodorization and bleaching to access emollient esters for the cosmetic market as desired white odorless product.13
Actual production costs depend very much on the available production infrastructure and company accounting standards. Thus, we cannot deliver cost estimates here. The largest contributors to cost are the choice of starting materials and the yield of the chemical transformation. Biocatalytic approaches may open entirely new avenues (see the Islatravir example, Chapter 6.1.3), which are found with a more open retrosynthetic view.10 Variable costs are further dependent on the amount of catalyst employed. Fixed costs largely depend on the space-time-yield (STY) of the transformation and the subsequent downstream processing. While the cost calculation will remain a case-by-case study, we intend to facilitate early on estimates by giving the following key performance indicators (KPIs) for as many examples as possible:
-
Yield (%) and/or selectivity/enantiomeric excess (%ee)
-
Substrate loading or product titer (g L−1 reactor volume)
-
Space-time-yield (STY, g L−1 h−1)
-
Catalyst consumption/load (i.e., g enzyme kg−1 product)
Looking at the sheer numbers, one can quickly appreciate that enzyme cost can range between single-digit cents kg−1 (for most efficient hydratase or isomerase processes) to several hundred € kg−1 (for some cytochrome P450 applications). The KPIs should hence help the process developer set targets and estimate the probability of success. Researchers at Codexis have exemplified this for the enzymatic reduction of a prochiral ketone (Table 2), which indicates the boundaries for a new process as well as the achievements made through enzyme engineering to finally reach these targets.
Parameter |
Desired value |
Initial process |
Final process |
|---|---|---|---|
Substrate loading [g L−1] |
>160 |
80 |
160 |
Reaction time [h] |
<10 |
24 |
8 |
Catalyst loading [g L−1] |
<1 |
9 |
0.9 |
Isolated yield [%] |
>90 |
85 |
95 |
STY[a] [g L−1 h−1] |
>16 |
3.3 |
20 |
- [a] STY, space-time-yield.
In the following chapters, we exemplify the most important developments for the use of biocatalysis in industrial applications for various target chemicals. The chapters are ordered by the key functionalities (alcohols, amines, carboxylic acids, etc.) created by enzymes as well as for glycosylations, more complex molecules and finally novel biocatalytic reactions which have the potential for industrial scale-up.
2 Alcohols
Chiral alcohols are important structural and functional motifs in many pharmaceuticals, fine chemicals and agrochemicals.15 In the pharma industry, building blocks with chiral hydroxyl moieties are key intermediates of multiple APIs of drug candidates.16 The rationale, and hence popularity, of accessing chiral alcohols by biocatalysis is obvious—stereocontrol in the synthesis, mild conditions, absence of metal-based catalysts and a reduced environmental footprint.
Kinetic resolution by lipases or asymmetric synthesis by KREDs are likely the most abundant enzymatic methods to make chiral alcohols and this will be illustrated in individual case studies below. The increasing popularity of these enzymatic reactions is reflected in a recent survey on patent activities, conducted in 2014–2019.17 One aspect when using KREDs is the stoichiometric requirement of the expensive cofactors NADH or NADPH. This can now be considered solved also on industrial scale as efficient recycling of NADH is simply performed using isopropanol as hydride donor without the requirement of a second enzyme, whereas NADPH recycling is commonly based on the use of a glucose dehydrogenase (GDH) where glucose serves as co-substrate. Hydroxylating enzymes will be discussed as an alternative.
2.1 Chiral Alcohols Produced by KREDs
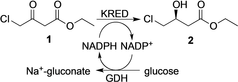
Dulox alcohol (S)-5 is a key precursor for the anti-depressant Duloxetine. BASF′s route takes advantage of the robust KREDs LbADH from Lactobacillus brevis18 and EbN1 from Aromatoleum aromaticum19 which both accept the labile chloro-ketone 3 and are highly selective for the reduction to the (S)-alcohol (S)-4.
An evolved enzyme proved fast and robust in mixed solvent systems.19b It demonstrates reduced product inhibition and accepts rac-2-butanol or isopropanol for the recycling of NADH. (S)-4 can then be easily aminated to afford (S)-5 (Scheme 1). An alternative route starting from the more stable dimethylammonium-ketone 6 can be achieved at 1 m concentration and near-perfect enantioselectivity with the KRED RtSCR9 from Rhodosporidium toruloides (Scheme 1).20 The primary shortcoming of this route is the subsequent N-demethylation of (S)-7. The use of glucose as terminal reductant has a low atom efficiency, but drives the equilibrium of the carbonyl reduction to completion—an advantage over the use of isopropanol, which even after prolonged distillative removal of acetone, struggles to reach quantitative conversion. The co-enzyme GDH is efficient and its consumption is usually insignificant compared to the lead KRED. A study comparing different ways of co-factor recycling and enzyme preparation has been published.21

A variety of alternative routes has been scouted by Novartis scientists to secure efficient and stereoselective access to LNP023, which is used as a treatment for patients with kidney disease caused by inflammation (further indications currently under review in clinical trials). One of the drawbacks of the previous synthesis route was the use of hazardous chemicals (such as sodium hydride, or dimethylacetamide representing safety concerns on a larger scale) and the poor enantio- and diastereoselectivity of the steps, leading to unwanted stereoisomers.
Enzymatic ketone reduction was introduced as a method to set one of the two stereocenters (Scheme 2).28 Compared with the prior routes, incorporation of this KRED step leads to a more efficient process, with full selectivity, convergency and easy execution.

Synthesis of the alcohol intermediate 10 of LNP023 by a KRED.28
Industrial processes in which a combination of different biocatalysts are used to set multiple chiral centers are described in the literature and clearly demonstrate the power of enzymatic processes (see also Chapter 6). One such example is the preparation of a gamma secretase inhibitor designed by Pfizer scientists using a transaminase for the synthesis of the key chiral amine building block 13 and a KRED for the reduction of an α-ketoester, delivering both fragments with high stereopurity (Scheme 3).29 The Pfizer team took advantage of commercially available enzymes for screening which provided confidence that the most successful hit would be available in suitable quantities for the multi-kg scale and with sufficient stability to be immediately applied. More applications of transaminases and their synthetic readiness are discussed in Chapter 3.1.

A route to the chiral intermediate 13 of a gamma-secretase inhibitor using a KRED and a transaminase.29
Recently, the Xu and Zheng group reported the directed evolution of KREDs for the synthesis of several important chiral alcohols with high STY, including an Imbruvica precursor (25 g L−1 h−1),31 an (R)-α-lipoic acid precursor (24 g L−1 h−1),32 and an Atorvastatin precursor 17 (44 g L−1 h−1, Scheme 4).30 For the production of 17, a route initially developed by Codexis was followed.14 A KRED (LbCR) from Lactobacillus brevis was subjected to directed evolution to improve thermostability and activity. Synergistic effects were found by combining the beneficial mutants, thus leading to LbCRM8 with 1900-fold increased half-life at 40 °C and a 3.2-fold increase in kcat/KM. Using E. coli cells (1 g CDW L−1) co-expressing this mutant and GDH, (5R)-16 (300 g L−1) was fully reduced to (3R, 5R)-17 in 6 h with a STY of 44 g L−1 h−1. Clearly, this KRED process is highly efficient for the industrial production of the chiral alcohol moiety for Atorvastatin.

An improved KRED process30 for the synthesis of t-butyl 6-cyano-(3R,5R)-dihydroxyhexanoate 17 for the production of Atorvastatin 18.
KREDs can be applied in desymmetrization as shown by Chen et al. for desymmetrization of ethyl secodione 19 to (13R,17S)-ethyl secol 20 (Scheme 5), which is a key chiral intermediate in the production of several steroidal drugs.33 The reduction is also highly demanding in regio- and stereoselectivity as only one keto function must be reduced and four diastereomers can be formed. Starting from an alcohol dehydrogenase (RasADH) from Ralstonia sp., several runs of directed evolution were performed to obtain a RasADH-F12 mutant with excellent selectivity towards (13R,17S)-20 and 183-fold activity compared to the wild-type. Using E. coli cells (20 g CWW L−1) co-expressing RasADH-F12 and GDH, 19 (20 g L−1) was fully converted to (13R,17S)-20 in 6 h on a 1 L scale. The enzyme and process are promising for further improvement for industrial implementation.

KRED-catalyzed desymmetrization of ethyl secodione 19 for the synthesis of intermediates for steroidal drugs.33
2.2 Chiral Alcohols Produced by Lipases
The lipase-catalyzed synthesis of chiral alcohols via kinetic resolution of racemates is well documented in multiple reviews, book chapters and even a whole book.2a, 3a-3i Since the discovery and development of KREDs for enantioselective reduction of ketones and KREDs availability from many commercial sources, lipase-mediated resolution lost its previous synthetic importance, unless the synthetic route gives opportunity for dynamic kinetic resolution, desymmetrization of prochiral material or if both enantiomers from kinetic resolutions are valuable products. For the purpose of this review, we selected a couple of examples to demonstrate that lipases still have their place in organic synthesis34 and are important biocatalysts especially for the regioselective synthesis of alcohols and in desymmetrization processes.
One of the rather recent processes using lipase is the selective acylation of the cyclopentene diol 21 to access a key intermediate of prostaglandins (Scheme 6). After optimization with commercial enzymes as catalysts, the best conversion and selectivity was achieved with lipase QL from Alcaligenes sp.26 The enzymatic process was carried out at the 200 kg scale of starting material. KPI parameters are summarized in Table 3, for comparison to other enzymatic reactions leading to chiral alcohols.

Selective mono-acylation of diol 21 by lipase QL.26
Enzyme |
Product |
Product conc. [g L−1] |
STY [g L−1 h−1] |
TTN (estim.)[a] |
Catalyst load [g kg−1 product][b] |
Ref. |
|---|---|---|---|---|---|---|
KREDs: |
|
|
|
|
|
|
EbN1 from Aromatoleum aromaticum |
(S)-4 |
62 |
8 |
40.000 |
13 (CDW) |
|
RtSCR9 from Rhodosporidium toruloides |
(S)-7 |
186 |
47 |
>20.000 |
54 (CDW) |
|
|
|
|
|
|
|
|
P450-monooxygenases: |
|
|
|
|
|
|
Beauveria bassiana |
HPOPS |
103 |
0.5 |
n. a. |
1 (CDW) |
|
recomb. Candida tropicalis |
Dodecane diacid |
150 |
1.4 |
n. a. |
100 (CDW) |
|
P450-BM3 var. in recomb. E. coli |
4-HO-isophorone |
6 |
1 |
18000 |
104 (CWW) |
|
P450-BM3 var. in recomb. E. coli |
5-HO-diclofenac |
3 |
0.6 |
2750 |
104 (CWW) |
|
|
|
|
|
|
|
|
Lipase: |
|
|
|
|
|
|
Lipase QL |
22 |
140 |
21.5 |
n.a. |
49 |
|
|
|
|
|
|
|
|
Oleate hydratase (OA): |
|
|
|
|
|
|
OA from Elizabethkingia meningoseptica in recomb. E. coli |
(R)-10-hydroxy-stearate |
100 |
4 |
n.a. |
103 (CFE) |
- [a] TTN, total turnover number; if no better data available: recombinant enzyme estimated to be 1/3 of CDW for E. coli fermentation [b] CDW: cell dry weight; CWW: cell wet weight; CFE, cell free extract.
2.3 Chiral Alcohols Produced by Enzymatic Hydroxylations
The need for robust, regio- and stereoselective hydroxylations in the synthesis of enantiomerically pure secondary, and especially tertiary alcohols, on industrial scale, remains unmet. One of the most prominent enzyme class for these monohydroxylations are cytochrome P450 monooxygenases. P450 and similar flavin-dependent monooxygenases show very useful selectivities for individual hydroxylation products that are unmatched by conventional chemical methods. Although reports of applications for whole-cell biotransformations of steroids, without identifying the responsible enzymes, were reported for the synthesis of pharmaceuticals many decades ago,35 only little progress has been made in implementing recombinantly expressed P450 enzymes on kilogram scale synthesis and beyond. Two examples of DSM/Innosyn are intensively optimized processes based on P450-BM3 mutants on the 100 L scale for the oxidation of α-isophorone 23 to the (R)-4-hydroxy isophorone 2424 and of diclofenac 25 to its 5-hydroxy-metabolite 2625a (Scheme 7). Despite careful process optimization, the consumption of biocatalyst remained too high (10 times more E. coli biomass than product) which disqualifies this oxidation methodology for fine chemical applications. Thus, the examples of successful transfer of monooxygenase-catalyzed hydroxylation of unactivated hydrocarbons remain very few: in the late 1990s BASF developed the p-hydroxylation of (R)-2-phenoxypropionic acid (POPS to HPOPS)22b, 36 and Cathay successfully installed the oxidation of alkanes and fatty acids to the diacids, mechanistically starting with a P450-mediated terminal hydroxylation (Chapter 4.2.3).23a, 23b Both processes rely on the metabolic network of naturally potent eukaryotic strains, leading to better efficiencies than all examples using P450 enzymes recombinantly expressed in E. coli (Table 3).

Hydroxylation of α-isophorone 23 or diclofenac 25 by a P450-monooxygenase from Bacillus megaterium expressed in E. coli.25a
There are reasons for the bad performance of P450s: while the usually poor expression level in E. coli may be overcome, the often observed low stability of the enzyme, as well as its complex mechanism (the catalytic cycle needs a reducing step requiring a coupled reductase and NAD(P)H prior to oxygen uptake) will always lead to slow turn-over and usually mediocre TTN.
An appealing alternative strategy, rather than trying to overcome P450s catalytic limitations, could be the development of other enzyme classes: peroxygenases and α-ketoglutarate dependent oxygenases. Peroxygenases carry a catalytic heme moiety similar to P450 monooxygenases and can also perform selective hydroxylations and other oxidations.[37] Peroxygenases consume hydrogen peroxide (H2O2) as a pre-reduced form of oxygen and do not need a co-reductant. Hence, turnovers can be an order of magnitude faster than P450-catalyzed oxidations. Dosing H2O2 as a liquid is easier and faster to accomplish than aeration in many production set-ups, and the higher cost of H2O2 compared to O2 is insignificant for fine chemical applications. Hence, peroxygenases have a much brighter perspective to become industrially relevant catalysts than P450s. Unfortunately, most research work has dealt with just one so-called “unspecific peroxygenase” (UPO) from Agrocybe aegerita which has good general robustness but a low tolerance for higher H2O2 concentration. In situ H2O2 generation systems are being developed to overcome this stability issue and to make peroxygenase more useful and applicable.38 These efforts are already reflected in higher TTN in comparison to P450s and with further enzyme discovery and protein engineering, this class of enzymes could become the next generation of hydroxylation biocatalyst.39
Another enzyme class for C−H activations are the α-ketoglutarate-dependent oxygenases. During catalysis, oxidative decomposition of α-ketoglutarate yields a high energy Fe(IV)-oxo intermediate that is capable of performing homolysis of unactivated C−H bonds, yielding a hydroxylated product following a radical recombination.40 The industrial application of these enzymes has been demonstrated in the hydroxylation of many amino acids.41 For example, a number of hydroxylases able to catalyze conversion of l-proline to all four monohydroxy isomers have been discovered. Some of the biocatalysts have been engineered to suit better large scale production of 4-hydroxy prolines.42 Examples were performed at 20–40 g L−1 of substrate. These reaction titers are already orders of magnitude higher than for a typical P450-catalyzed reaction.
Another interesting enzyme class to form hydroxyl groups are hydratases, which add water to double bonds.43 The most prominent examples are the flavin-dependent oleate hydratases (OA), which convert oleic acid in a regio- and stereoselective fashion into the corresponding (R)-10-hydroxystearic acid and product formations of up to 100 g L−1 have been reported (Table 3).27 The high activity of OAs also enabled cascade reactions to afford functionalized fatty acid derivatives including long chain aliphatic amines.44 Only a few years ago, the first structure of an oleate hydratase was solved for the OA from Elizabethkingia meningoseptica45 that provided valuable insights into the mechanism of these enzymes. Moreover, this created the basis for protein engineering to enable asymmetric hydration of various terminal and internal alkenes, whereas the wildtype enzyme requires the presence of the carboxylic group function in fatty acids. This was overcome by using a carboxylic acid decoy molecule for activation of the enzyme from E. meningoseptica. Thus, the asymmetric hydration of unactivated alkenes was achieved on preparative scale with up to 93 % conversion at excellent selectivity (>99 %ee, >95 % regioselectivity).46
3 Amines
Chiral amines are of great importance in the pharmaceutical and agrochemical industry. More than 90% of current top-selling or newly approved small molecule drugs are amines or originate from amines. Most of them are chiral, and about 30 % of crop protection actives are chiral amine molecules.47 Optically pure amines therefore have a special focus in biocatalysis.48
3.1 Optically Active Amines
The most versatile biocatalytic approach to primary amines is the transaminase reaction that converts carbonyl substrates in a reductive amination reaction to the target amine. Concomitantly, it requires a sacrificial amine donating source (Scheme 8). The full scope has been reviewed extensively48b, 49 and tribute to its application on large-scale chemistry has been given.48a, 49b

Transaminase-catalyzed reductive amination exemplified for (S)-Moipa 28.
The use of the pyridoxal-5′-phosphate (PLP)-dependent transaminases for preparative synthesis of enantiomerically pure compounds has been pioneered by Celgene (later “Celgro”). Celgene initially applied the reversible transaminase reaction in a de-amination mode to deracemize or polish chiral amines (e.g. 1-phenyl-3-aminobutane and 1-arylethylamines).50 While the resolution worked well up to the scale of 160 L, the use of expensive amine acceptors like pyruvate or oxaloacetic acid hindered industrial application. The breakthrough for synthetic application came with Celgene‘s discovery of the particular advantages of isopropylamine as amine donor.51 Celgene showed preparative usefulness of the transaminase technology for enantiopure l-alanine (from pyruvate) and (S)-Moipa ((S)-1-methoxy-isopropylamine, 28; Scheme 8) and (S)-2-amino-3-methylbutane from the respective ketones. The latter amines serve as precursors for herbizides. Several thousand tons of (S)-Moipa are produced annually as building block for (S)-Dimethenamide 29 and could potentially serve for (S)-Metolachlor 30 (Scheme 9). A particularly remarkable achievement of Celgene's development was to overcome product inhibition by enzyme engineering.52 Eventually, Celgene could make (S)-Moipa to almost 2 m concentration (Table 4). Nonetheless, transaminase technology could not quite compete with lipase technology for (S)-Moipa or with an intensely optimized Ir-catalyzed imine hydrogenation route to (S)-Metolachlor.53

(S)-Dimethenamide 29 (BASF) and (S)-Metolachlor 30 (Syngenta).
Technology |
Product |
Product conc. [g L−1] |
STY [g L−1 h−1] |
TTN (estim.) |
Catalyst load [g kg−1 product] |
Ref. |
|---|---|---|---|---|---|---|
Crystallization of diasteromeric salts |
(R)- or (S)-1-PEA 31 |
50 (0.4 m) |
low |
– |
n.d. (90–95 % recovery of mandelic acid reported) |
|
Lipase |
1-PEA 31 |
(neat) |
>1000 |
107 |
<0.5 (immob Enzyme) |
|
Transaminase |
(S)-1-PEA (94 % conv.) |
6 |
1 |
103 |
800 (dry CFE) |
|
Transaminase |
(R)-1-PEA (80 % conv.) |
40 |
2 |
– |
125 (dry CFE) |
|
Transaminase |
(R)- or (S)-1-PEA (>90 % conv.) |
50 |
3 |
104 |
100 (dry CFE) |
|
Transaminase |
l-Alanine |
90 (1 m) |
5 |
– |
50 (wet cells) |
|
Transaminase |
(S)-Moipa 28 |
170 (2 m) |
25 |
105 |
20 CDW |
|
Transaminase |
36 |
156 |
7.8 |
– |
20 (lyophilized CFE) |
|
Lipase |
(S)-Moipa 28 |
(neat) |
300 |
107 |
<1.0 (immob Enzyme) |
|
Transaminase |
Sitagliptin 34 |
190 |
8 |
25000 |
32 (dry CFE) |
|
Rh-cat enamine hydrogenation |
Sitagliptin 34 |
110 |
7 |
670 |
3 (Rh-cat with chiral ligand) |
|
RedAm |
42 |
35 |
9 |
– |
8 CDW |
|
Aspartase (lyase) |
Aspartate |
166 |
140 |
– |
<0.5 (immob E. coli) |
- [a] 1-PEA, 1-phenylethylamine; RedAm, reductive aminase; STY, space-time-yield; TTN, total turnover number; CDW, cell dry weight; CFE, cell free extract.
The reason for the success of lipase technology for the resolution of simple chiral amines is the extraordinarily high activity of some lipases. Burkholderia plantarii lipase (BPL) and Candida antarctica lipase B (CALB) immobilized on polyester resin very selectively acylate the (R)-enantiomer (Scheme 10—exemplified for 1-phenylethylamine, 1-PEA, 31). Researchers at BASF and shortly later at Bayer noticed the high acceleration of the lipase catalysis when methoxyacetic acid esters were used as acylation reagents, a breakthrough for this technology.54

Lipase-catalyzed kinetic resolution of racemic benzylamines exemplified for 1-phenylethylamine 31.55
The full scope of the lipase technology has been reviewed.55 For alkylbenzylamines, the (R)-enantiomer gets acylated with often very high selectivity (E>1000), so that at 50 % conversion of the racemate essentially both, (S)-amine and (R)-amide can be recovered at 99 %ee after distillative separation. (S)-Moipa, other substituted aminoalcohols, and a large number of chiral alkyl-benzylamines are now being separated with lipase technology on a several-thousand-tons per annum scale.
Biocatalytic transamination and lipase technologies both show advantages compared to classical resolution of enantiomers by crystallization of diastereomeric salts. Most importantly: solid handling, i.e., centrifugation or particularly expensive filtration, are avoided—a big advantage for larger volume manufacturing. Products and by-products can be separated by distillation or extraction. The (S)-amide can be saponified without loss in optical purity.56 In some cases, the lipase technology can be run entirely without solvent which further reduces recycling loops and leads to very high STY (Table 4). In addition, the acylation reagent is recycled57 leading to minimal waste (only stoichiometric amounts of caustic soda and sulfuric acid are consumed). A frequently encountered disadvantage of racemate resolution, the loss of the less demanded enantiomer, has been solved in the BASF process as it can be returned to the initial (reductive) amination process which runs under racemizing conditions.58
Transaminase technology has found its successful application in the syntheses of more complex pharma intermediates, the most famous example being the synthesis of the diabetes drug Sitagliptin 34 by Merck & Co. from the prochiral precursor pro-Sitagliptin 33 (Scheme 11).59 Starting from ATA-117, a close homologue of the wild-type enzyme, which had no detectable activity on the substrate, the first variant provided very low activity (0.2 % conversion of 2 g L−1 substrate using 10 g L−1 enzyme) towards pro-Sitagliptin. The final variant created by several rounds of directed evolution converts 200 g L−1 ketone to Sitagliptin with excellent selectivity (99.95 %ee) at 92 % yield using simply isopropylamine as amine donor. Compared to the previously used chemical process with a Rh-t-Bu-Josiphos catalyst for asymmetric hydrogenation at high pressure, the enzymatic route resulted in higher overall yield, 53 % higher productivity, reduced total waste and elimination of the transition metal catalyst.60

Asymmetric synthesis of Sitagliptin 34 using an engineered transaminase (ATA).59
As in the sitagliptin example, similar reasons led scientists at Novartis to explore novel routes for the synthesis of Sacubitril, one of the two active pharmaceutical ingredients of the supramolecular complex LCZ696, an angiotensin receptor neprilysin inhibitor.61 The design of the novel route for the synthesis of Sacubitril was dictated by the potential for improved efficiency and green metrics—attributes which enzymes can affect significantly. The favorite retrosynthetic approach was a route using a transaminase for installation of a chiral amine functionality starting from the corresponding γ-keto acid 35 (Scheme 12). Also in this case, the enzyme had to be evolved in multiple rounds, with a total 500,000 fold improvement over the parent enzyme62 to accept the sterically challenging substrate and to perform the reaction at elevated temperature (58 °C). The transaminase process was introduced at commercial scale and delivered the key intermediate 36 for the synthesis of a blockbuster for cardiovascular treatment (for KPI parameters see Table 4), with reduction of the carbon footprint by factor 3.63

Transaminase-catalyzed asymmetric synthesis of an intermediate of a blockbuster for cardiovascular treatment.63a
Despite the demonstrated robustness and versatility of transaminases in the preparation of chiral amines on industrial scale, the obvious downside is that only primary amines can be prepared by this enzyme class.
Chiral amines (primary, secondary, tertiary) can be accessed via a chemo-enzymatic approach: monoamine oxidases (MAOs)-catalyzed enantioselective amine oxidation to imines and simultaneous non-selective chemical imine reduction.71 The Turner group pioneered MAO-N from Aspergillus niger for the synthesis of several bulky APIs and natural products72 including a chiral secondary amine intermediate for the drug Boceprevir by Merck & Co. and Codexis.73
Accessing secondary and tertiary amines can be achieved by imine reductases (IREDs) catalyzing reduction of a C=N bond with stoichiometric consumption of NADPH. IRED activity was first described in whole-cell systems74 for stable, aromatic or cyclic imines with low titer and STY (8 g L−1 in 84 h for (R)-2-methylpyrrolidine), and then identified as an NADPH consuming enzyme class with specific activity of 3 U mg−1.75 The synthetic use of IREDs has remained limited to cyclic imines, some aromatic imines which are stable in water,76 and heterocyclic imines.77 We are aware of only one example of enzymatic imine reduction on larger scale. Pfizer scientists use IREDs to get access to (S,S)-Sertraline 39 from the corresponding imine 38 (Scheme 13).78

IRED-mediated imine reduction for the synthesis of Sertraline 39.78
A far brighter perspective to imine reduction came with the discovery of structurally related reductive aminases (RedAms). Although Merck & Co. already had developed an efficient asymmetric synthesis route using a transaminase79 to make the drug Vernakalant, an antiarrhythmic agent, they also explored the use of IREDs. Starting from an opine dehydrogenase from Arthrobacter sp., eleven rounds of directed evolution afforded a variant with 29 mutations resulting in the desired product with 80 %de, notably the first example of an engineered “reductive aminase” (RedAm).80 This was then followed by Aleku and Turner with the discovery of the AspRedAm reductive aminase.81 This reductive aminase is highly homologous to IREDs and is similarly NADPH-dependent. These enzymes can catalyze imine formation simultaneously to its reduction, thus effectively making reductive amination possible.82
The full synthetic potential of RedAms became apparent in recent work by GSK in the process development towards an LSD1 inhibitor (GSK2879552).69 Only three rounds of evolution were necessary to obtain a more active, more stable, and pH-adjusted RedAm-variant for the resolution of racemic trans-tranylcypromine 40 in the reductive amination with aldehyde 41 to obtain the target intermediate 42 (Scheme 14) in 84 % yield and excellent chemical (99.9 %) and optical purity (99.7 %ee). Compared to the previously executed conventional route, green metrics were greatly improved. Product titers, STY and catalyst consumption play in the same league as the best transaminases (Table 4). The RedAm catalyzes the key step in a very convergent synthesis, making this transformation particularly valuable.

Enzymatic reductive amination for the synthesis of the key intermediate 42 of the LSD1 inhibitor GSK2879552.69
3.2 Achiral and Racemic Amines
While biocatalysis has developed to the leading technology for the manufacturing of enantiomerically pure chiral amines, the by far largest volumes (and values) are achiral or racemic amines. Conventionally, alcohols and carbonyls are reacted with excess NH3 under H2-atmosphere at elevated temperature (150–250 °C) on a heterogenous transition metal catalyst (mostly Cu, Ni, Co on oxide support).83 Typically, STY of 0.1–1 kg L−1 h−1 are achieved in gas-phase or liquid phase modus.
The relatively low activity of the metal catalyst and the high temperatures needed limit the scope of the heterogeneous amination. In recent years, biocatalytic approaches have succeeded to mimic the (redox neutral) alcohol amination, either by a three-enzyme system based on an alcohol dehydrogenase, a transaminase and an alanine dehydrogenase)84 or using a two-enzyme system85 based on a mutated amino acid dehydrogenase.86 Alternatively, fatty amines were produced from fatty acids in a one-pot tandem cascade using a combination of a carboxylic acid reductase (see Chapter 7) and a transaminase in up to 96 % conversion.87 However, the total activity of the combined enzyme systems has so far remained too low to reach technical applicability.
A review on the utilization of these enzymatic transformations in metabolically engineered microorganisms for amine derivatives has been published.88 Performance parameters for these whole-cell approaches are hardly encouraging. Nonetheless, Evonik has scaled the synthesis of ω-aminolauric acid from lauric acid in a whole-cell catalyst (comprising enzymatic oxidation and transamination) into the pilot scale.89
3.3 Amino Acid Production by Lyases
Processes for the synthesis of amino acids using lyases have been summarized in reviews and publications.2a, 70, 90 Adding ammonia to fumarate with aspartase is the preferred method to make l-aspartic acid, not just of interest as amino acid itself, but also as precursor for the sweetener Aspartame. The subsequent enzymatic decarboxylation of l-aspartate to l-alanine catalyzed by an l-aspartate-β-decarboxylase is still a viable industrial route to l-alanine, despite the obvious atom inefficiency. The l-aspartate-α-decarboxylase is useful to make β-alanine from aspartate.91a
The naturally highly specific aspartase was opened to promiscuity for other substrates by Vogel et al.91b using conventional enzyme engineering and then by Janssen and Wu et al.91c with computational redesign to access several chiral β-amino acids with excellent regio- and enantioselectivity in up to 300 g L−1.91c
EDDS-lyase is a related enzyme that degrades (or synthesizes) the strong natural chelator and siderophore (S,S)-ethylenediamine disuccinate (S,S-EDDS). Beyond its high natural reactivity, EDDS-lyase is of great interest for synthetic chemists as it shows a good promiscuity in the choice of the amine donor. The group of Poelarends has elucidated the structural basis for this promiscuity92 and engineered mutants that open new synthetic approaches to the Aspartame related sweeteners Neotame and Advantame.93
An interesting example for the combination of biocatalysis and homogeneous catalysis is the synthesis of (S)-2-indolinecarboxylic acid 45 (Scheme 15), a key intermediate in the production of angiotensin 1-converting enzyme inhibitors such as Indolapril and Perindopril. DSM developed a route using a phenylalanine ammonia lyase (PAL) from Rhodotorula glutinis using whole cells with the recombinant PAL expressed in E. coli.94 The amino acid intermediate 44 was obtained at 91 % yield with 99 %ee. Optimization of the subsequent copper-catalyzed ring closure enabled to use 4 mol % CuCl to obtain the final product after just 4 h in 95 % yield and 99 %ee. Notably, the bromo derivative underwent faster ring closure at only 0.01 mol % CuCl, but the bromo-derivative was a less good substrate in the PAL-catalyzed step. This process has been scaled up by DSM for ton scale production. A life cycle analysis revealed that the carbon footprint of this process is reduced by half compared to an older process, mostly because of substantially reduced usage of organic solvents.

A combination of an enzymatic reaction catalyzed by PAL with copper-catalyzed ring closure affords a key intermediate for angiotensin 1-converting enzyme inhibitors.94
One of further examples is the use of an engineered PAL for hydroamination of the cinnamic acid derivative 46 to the corresponding phenylalanine analogue 47 (Scheme 16) which is then telescoped to a Pictet-Spengler reaction with formaldehyde. The corresponding tetrahydroisoquinoline derivative 48 was isolated in 60–70 % yield and with >99.9 %ee.95 In this case, the enzyme was engineered for enhanced activity and especially stability at high pH (9.5–10.5) and high concentration of ammonia (9–10 m) to push the reaction equilibrium and to maximize yield. After careful optimization, this process—scaled up to 2 kg at Novartis—was used for the preparation of EMA401, an angiotensin II type 2 antagonist for treatment of postherpetic neuralgia and neuropathic pain. This is a more sustainable, shorter and more cost-effective alternative to the previous process.

PAL-mediated synthesis of the key intermediate 47 for the production of EMA401.95
4 Carbonyls, Carboxylic Acids and Derivatives
4.1 Carbonyls
Aldehydes and ketones are common organic chemicals that are generally produced via chemical processes. However, biocatalytic methods may provide several advantages, such as customers’ preference of “all-natural production” in flavors and fragrances and high regioselectivity to produce ketoses. In general, alcohol oxidation by dehydrogenases or oxidases96 and C−C bond formation by aldolases or lyases97 are the two main approaches for efficient production of aldehydes and ketones.
Recently, the Hollmann group reported a highly efficient alcohol oxidation process to produce trans-2-hexen-1-al, a green note aroma (50, Scheme 17).98 An aryl alcohol oxidase from Pleurotus eryngii (PeAAOx, 0.75 μm) and a catalase (0.1 μm) were applied to convert 49 (500 mm) in a two-liquid-phase system. Full conversion to 50 (49 g L−1) was achieved within 24 h. Furthermore, the oxidation process could run with pure 49 as the organic phase: 255 g L−1 of 50 was accumulated after 14-days with multiple additions of PeAAOx and catalase. Although the conversion is not complete (31 %), the TTN of PeAAOx reached 2.2×106. This study proves the very high catalytic efficiency of alcohol oxidases and demonstrates the potential for industrial production of aldehydes. For broad-scope oxidation, the Turner group recently engineered a choline oxidase to accept a wide range of primary alcohols.99 Although the activity is not very high (<1 U mg−1), the broad scope offers an excellent start for laboratory syntheses and directed evolution for industrial applications.

Oxidation of trans-2-hexen-1-ol 49 to trans-2-hexen-1-al 50 by an aryl alcohol oxidase.98
Galactose oxidase (GalOx) is a well-known, efficient copper-based alcohol oxidase which shows some promiscuity beyond its natural sugar substrates.101 Its oxidation with O2 can usually be directed quite selectively to the aldehyde, but oxidation can go on to the acid if the aldehyde readily forms hydrates. A TTN of 106 has been achieved, qualifying GalOx as a suitable oxidase for large scale oxidation. A recent example is the oxidation of 5-hydroxymethylfurfural (51) to diformylfuran (52) (Scheme 18).100 Almost quantitative yield can be achieved with decent STY (4 g L−1 h−1) at low enzyme load (2 g kg−1 of product). 52 can be further oxidized to the monomer 2,5-furan dicarboxylic acid (Chapter 4.2.3).

Oxidation of hydroxymethylfurfural to diformylfuran by galactose oxidase.100
A common problem for biocatalytic production of aldehydes is the susceptibility for reduction/oxidation by native dehydrogenases in microbial cells.103 Recently, the Xu group developed a facile solution by co-expressing thermophilic enzymes in E. coli and performing the cascade conversion of the lignin-derivative ferulic acid (53) into vanillin (55, Scheme 19) at elevated temperatures.102, 104 Two thermophilic enzymes were identified: a phenolic acid decarboxylase (PAD) from Bacillus coagulans DSM1 and an aromatic dioxygenase (ADO) from Thermothelomyces thermophila. Conversion of 53 (100 mm) with E. coli co-expressing PAD and ADO produced 55 (87 mm, 13.3 g L−1) in 18 h at 50 °C and pH 9.5. This biocatalytic process has the potential for industrial application because this route (bio-based substrates and enzyme cascade) matches the customers’ preference for bio-vanillin production.

Cascade conversion of ferulic acid 53 into vanillin 55 with thermophilic enzymes.102
Due to high regioselectivity, enzymes are particularly suitable for oxidation of polyols and sugars to produce polyhydroxylated ketones and ketoses.105, 106 The Chaiyen group reported the production of l-ribulose from l-arabinose by combining an engineered pyranose 2-oxidase, xylose reductase, and assisting enzymes.106 For practical synthesis, the Ouyang group reported efficient oxidation of galactitol (56) to d-tagatose (57) by a robust polyol dehydrogenase (PdPDH) and an NADH oxidase (StNOX, Scheme 20).105 The biocatalytic process was performed on a 2 L scale using two lyophilized E. coli cells (total: 9.7 g) containing PdPDH and StNOX, respectively. 56 (200 g) was fully converted to 57 in 15 h with a STY of 6.7 g L−1 h−1. Further work-up offered pure 57 in 91 % yield. This study demonstrated the potential of biocatalytic oxidation of polyols to produce high-value ketoses.

Oxidation of galactitol 56 to d-tagatose 57 by a polyol dehydrogenase.105
Besides oxidation, polyhydroxylated ketones and ketoses could be produced via C−C bond formation by aldolases or lyases.114, 115 Fessner and Clapes engineered fructose-6-phosphate aldolase (FSA) from E. coli to convert alkanones and alkanals into chiral β-hydroxyl ketones/aldehydes.115a FSA was also combined with a transketolase and a transaminase for sequential one-pot synthesis of l-ribulose, d-tagatose, and l-psicose.115b For a practical synthesis, the Sun group utilized FSA(A129S) in E. coli cells (30 g CDW L−1) to convert formaldehyde (58, 3 m) and dihydroxyacetone (59, 3 m) into l-erythrulose (60, Scheme 21).114 The target compound 60 (2.21 m, 252 g L−1) was produced within 2 h with a STY of 126 g L−1 h−1, showing the potential of aldolases for industrial production of ketoses.

Conversion of formaldehyde and dihydroxyacetone to L-erythrulose 60 by an aldolase.114
Deoxyribose phosphate aldolase (DERA) has been used in several variations for industrial production, in particular for the statin side chains. DERA′s impressive applications have recently been reviewed.116
4.2 Carboxylic Acids and Esters
Carboxylic acids are widely used in the chemical, food, material, and pharmaceutical industries. Biocatalysis is most suitable for industrial production of high-value (chiral) carboxylic acids (Table 5). Due to the recent advances in directed evolution and enzyme cascades,117 biocatalysis has shown potential at laboratory scale for the production of bulk carboxylic acids.
Enzyme |
Product |
Product conc. [g L−1] |
STY [g L−1 h−1] |
TTN (estim.)[a] |
Catalyst load [g kg−1 product] |
Ref. |
|---|---|---|---|---|---|---|
Nitrilase: |
|
|
|
|
|
|
BCJ2315 from Burkholderia cenocepacia J2315 |
(R)-63 a |
350 |
15 |
60.000 |
11 (CDW[b]) |
|
Nitrilase (A190 H) from a metagenomic library |
(R)-66 |
390 |
26 |
11.000 |
26 |
|
Nitrilase mutant from Acidovorax facilis ZJB09122 |
74 |
220 |
37 |
10.000 |
3.3 (CDW) |
|
|
|
|
|
|
|
|
Hydrolase: |
|
|
|
|
|
|
Lipase from Thermomyces lanuginosus (TLL) |
(S)-77 |
320 |
13.5 |
10.000 |
15 |
|
Protease C from Bacillus subtilis |
(R)-79 |
87 |
4.6 |
n.a. |
240 |
|
Lipase B (I189K) from Candida antarctica (CALB) |
(2R, 3S)-81 |
120 |
24 |
170.000 |
0.83 |
|
|
|
|
|
|
|
|
Hydroxynitrile lyases: |
|
|
|
|
|
|
PaHNL(A111G) from Prunus amygdalus |
(R)-62 b |
360 |
60 |
200.000 |
0.2 |
- [a] TTN, total turnover number; if no better data available: recombinant enzyme estimated to be 1/3 of CDW for E. coli fermentation [b]CDW: cell dry weight.
4.2.1 Acid Production via Hydrolysis of Nitriles
One of the most efficient biocatalytic approaches for carboxylic acids is hydrolysis of nitriles by nitrilases or nitrile hydratase-amidase system, because these enzymes are highly active without the requirement for external cofactors.118 Nitrilase-based processes have been implemented in industry on the multi-ton scale for more than 20 years, such as the production of nicotinic acid (Lonza)119 and (R)-mandelic acid ((R)-63 a, Scheme 22 a) (BASF and Mitsubishi Rayon).2a

The process to produce (R)-mandelic acids is an elegant dynamic kinetic resolution due to the in situ racemization of cyanohydrins and offers the final products in 100 % theoretical yield (Scheme 22 a).107, 120 One of the most productive processes has been reported by the Wei group using E. coli cells expressing a nitrilase (BCJ2315) from Burkholderia cenocepacia J2315.107 By applying a continuous feeding of mandelonitrile 62 a, (R)-63 a was produced at a concentration of 2.3 m (350 g L−1) with 97.4 %ee in 24 h on 10 L scale. After further work-up, (R)-63 a was isolated in 93 % yield and 99.5 %ee. The high STY (15 g L−1 h−1) and low biocatalysts loading (3.9 g CDW L−1) demonstrate the potential for industrial implementation. (R)-o-Chloromandelic acid ((R)-63 b) is a key intermediate for the antiplatelet drug (S)-Clopidogrel 64. However, due to the steric hindrance of the ortho-chloro substituent, most of the native nitrilases are inferior in terms of activity and/or enantioselectivity. A double mutant (I113M/Y199G) of the nitrilase BCJ2315 was engineered with higher activity and enantioselectivity.120 Using E. coli cells expressing this mutant (3.9 g CDW L−1), (R)-63 b was produced at a concentration of 500 mm (93 g L−1) with 98.7 %ee in 3 h. The STY reaches 31 g L−1 h−1 (Table 5).
A related process for the production of α-hydroxy acids from aldehydes is based on hydroxynitrile lyases (HNL), which catalyze the asymmetric addition of HCN to carbonyls.122 HNL processes have been applied in industry for manufacturing of chiral mandelic acids for about 20 years by DSM and other companies (Scheme 22 b).2a Glieder et al. applied protein engineering to tailor PaHNL from Prunus amygdalus (Almond) for highly efficient synthesis of (R)-63 b.113, 121a Importantly, PaHNL is stable under acidic conditions with minimal racemization of cyanohydrins. PaHNL variant A111G was identified with very high activity (409 U mg−1) towards 61 b.113 Biotransformation of 61 b (3 m) with this variant (0.1 g L−1) produced (R)-62 b (97 %ee) with 96 % isolated yield in 7 h. The STY reaches 60 g L−1 h−1 and the product/catalyst ratio is more than 4000. Further evolving PaHNL led to variants with even higher activity and enantioselectivity.121a Asano et al. have recently described very active (8 kU mg−1), robust and enantioselective HNLs from millipedes for (R)-62 a synthesis121b and have shown how to engineer these for (R)-62 b.121c The HNL process shows favorable performance data for the synthesis of (R)-63 b.
Another elegant example of nitrilase is the desymmetrization of 3-hydroxyglutaronitrile (65) to give (R)-4-cyano-3-hydroxybutyrate ((R)-66) for the synthesis of Atorvastatin as developed by Diversa (Scheme 23 a).108, 123 Initially, a nitrilase from a metagenomic library produced (R)-66 in 98 % yield yet with reduced enantioselectivity with high substrate loading.123a Directed evolution was applied to identify an A190G mutant with higher enantioselectivity.108 With this improved nitrilase (10 g L−1) and high loading of 65 (3 m, 330 g L−1), (R)-66 was produced in 96 % yield 98.5 %ee in 15 h with a STY of 26 g L−1 h−1.123b Recently, the Zhu and Wu group reported desymmetrization of 3-substituted glutaronitriles followed by Curtius rearrangement and hydrolysis to prepare (S)-Pregabalin and (R)-Baclofen 69 (Scheme 23 b).124 For practical synthesis, they further engineered a nitrilase from Synechocystis sp. PCC6803, and the best variant (P194A/I201A/F202V) showed 11 times higher activity towards 67 and excellent enantioselectivity.124b Preparation of (S)-68 in 91 % yield and 99 %ee was achieved by hydrolysis of 67 (100 mm) in 1 h with E. coli cells expressing the nitrilase variant. The STY reaches 20 g L−1 h−1 (Table 5).

Nitrilase can be applied for kinetic resolution to produce chiral carboxylic acids, as shown in a recent process to produce Pregabalin (Scheme 24).125 Highly regio- and enantioselective hydrolysis of isobutylsuccinonitrile (70, 100 g L−1) with a nitrilase from Brassica rapa produced (S)-3-cyano-5-methylhexanoic acid ((S)-71) in 47.5 % conversion and 98 %ee in 3 h.125a Noteworthily, the catalysts, immobilized E. coli cells, facilitated the downstream process and were re-used for 12 batches while maintaining performance (>41.1 % conversion, >98 %ee). The unreacted (R)-70 was isolated and racemized to starting material, while (S)-71 was easily hydrogenated to Pregabalin 72, offering much less waste compared to other routes. Based on this nitrilase, the same group further engineered a hybrid nitrilase with higher activity and excellent enantioselectivity for (S)-71: an even more practical synthesis of Pregabalin can be therefore expected.125b

A nitrilase process (kinetic resolution) for the synthesis of (S)-3-cyano-5-methylhexanoic acid 71 for the production of Pregabalin, 72.125
Regioselective hydrolysis of dinitriles by nitrilases can also provide useful achiral carboxylic acids, such as 1-cyanocyclohexane acetic acid (74), an intermediate for Gabapentin 75 (Scheme 25). The Zheng group engineered a mutant of a nitrilase from Acidovorax facilis ZJB09122 with improved activity, thermostability, and product tolerance.109 Under the optimal conditions, 1.5 m 1-cyanocyclohexylacetonitrile (73, 222 g L−1) was completely hydrolyzed by E. coli expressing the nitrilase (14 g CDW L−1) in 6 h with a STY of 37 g L−1 h−1 (Table 5). 74 was easily isolated in 88 % yield. Besides these well-developed drugs, nitrilases have been widely applied in the preparation of (chiral) carboxylic acids/nitriles for many drug candidates in development.17, 126 Regio- and enantioselective hydrolysis of nitriles by nitrilases is thus a highly efficient and mature approach for industrial production of (chiral) carboxylic acids.

A nitrilase process (regioselective hydrolysis) for the synthesis of 1-cyanocyclohexane acetic acid 74 for the production of Gabapentin 75.109
4.2.2 Acid Production via Hydrolysis of Esters/Amides
Hydrolysis of esters and amides by esterase or lipase has been well-established for industrial production of chiral carboxylic acids for more than 20 years.127 Currently, many lipases and esterases are commercially available, stable in organic solvents, and with high enantioselectivity for (dynamic) kinetic resolution or desymmetrization. Thus, they have been widely applied for the production of chiral acids as synthetic intermediates for pharmaceuticals.128
For the production of Pregabalin, a team from Pfizer developed a second-generation process based on lipase-catalyzed resolution (Scheme 26).110 The commercial Thermomyces lanuginosus lipase (TLL) showed very high (S)-enantioselectivity (E>200) for hydrolysis of diester 76. In the optimized process, 76 (765 g L−1) was hydrolyzed by TLL (12 %) to give (S)-77 with 47.5 % conversion in 24 h. The STY reaches 13.5 g L−1 h−1. The process had been scaled up in manufacturing trials at 3.5 tons (8000 L reactor). However, due to the moderate activity of TLL, the enzyme loading is still too high. Recently, the Zheng group engineered a TLL(S58L/S83T) mutant showing 5.5-fold higher specific activity than the wild-type TLL.129 By applying E. coli whole cells (5 % w/v) expressing this TLL mutant, 3 m 76 (765 g L−1) was hydrolyzed to (S)-77 (96 %ee) with 45 % conversion in 24 h (Table 5).

A team from USB Pharma developed a hydrolase-based process for the synthesis of the (R)-succinic acid derivative (R)-79 for the production of Brivaracetam (Scheme 27).111 A protease C from Bacillus subtilis was identified for enantioselective hydrolysis of racemic ester 78. Hydrolysis of 78 (1 kg) was achieved with the protease C (10 %, w/w) in water (4.5 L) to give (R)-79 (97 %ee) with 42 % isolated yield in 19 h. The STY is about 4.6 g L−1 h−1, but the loading of enzyme (10 %, w/w) is too high for industrial application.

A hydrolase-based kinetic resolution process for the synthesis of the (R)-succinic acid derivative 79 for the production of Brivaracetam 80.111
The power of directed evolution of lipase was well-demonstrated in a recent report about the engineering of the Candida antarctica lipase B (CALB) for the resolution of cis-dimethyl-1-acetylpiperidine-2,3-dicarboxylate (cis-81) for the production of Moxifloxacin 82 (Scheme 28).112 A single I189K mutation of CALB boosted its specific activity more than 200-fold while maintaining the excellent enantioselectivity in the hydrolysis of cis-81. With the purified CALB variant (I189K) (0.1 g L−1), 1 m cis-81 (243 g L−1) was hydrolyzed to give (2S, 3R)-81 with 49.9 % conversion in 5 h. The STY reaches 24 g L−1 h−1 with a high product/catalyst ratio of 1200. Furthermore, this CALB variant (I189K) has been immobilized and applied for the resolution of cis-81 (100 g L−1) in a stirred tank reactor for 50 cycles (average STY: 50 g L−1 h−1) or in a recirculating packed bed reactor for 50 cycles (average STY: 59 g L−1 h−1).130

4.2.3 Acid Production via Oxidation of Alcohols/Alka(e)nes
Carboxylic acids can be potentially produced by oxidation of readily available alcohols or alkanes. Enzymatic oxidation often utilizes molecular oxygen as “green” oxidant and takes place under mild conditions, thus being a greener alternative to chemical oxidations.39, 131 In comparison to hydrolysis, enzymatic oxidation is often less efficient, but with great potential due to advances in enzyme discovery and directed evolution. Biocatalytic oxidation has been employed in the food industry for oxidation of polyols and sugars to their corresponding acids, such as oxidation of glucose to d-gluconic acid by glucose oxidase.2a
A recent practical process is the selective oxidation of glycerol (83) to d-glyceric acid ((R)-84) by acetic acid bacteria (Scheme 29). When growing Gluconobacter frateurii NBRC103465 in a medium with high initial glycerol concentration (170 g L−1), 137 g L−1 of (R)-84 (72 %ee) was accumulated in the culture broth in 6 days.132 Using Acetobacter tropicalis NBRC16470 in a medium loaded with glycerol at the outset (220 g L−1), 102 g L−1 of (R)-84 (99 %ee) was accumulated over 6 days. The processes took much longer than a usual biocatalytic process because of the time required for strain cultivation.

Selective oxidation (desymmetrization) of glycerol 83 to d-glyceric acid 84 by whole cells of acetic acid bacteria.132
A very attractive reaction is the oxidation of 5-hydroxymethylfurfural (85) to 2,5-furan dicarboxylic acid (86, Scheme 30) as a building block for biobased polymers.134 Many enzymes and enzyme systems have been reported, yet most of them suffer from limited efficiency and relatively low substrate concentrations (≤100 mm, 16 g L−1).133a-133c Using whole cells may provide a productive process.133d-133g Koopman and Wierckx engineered a recombinant Pseudomonas putida S12 strain co-expressing an HMF/furfural oxidoreductase, a transporter and an aldehyde dehydrogenase.133d, 133e By using fed-batch cultivation of the engineered strain, more than 150 g L−1 of 86 was produced from biotransformation of 85 in about 92 h. It will be interesting to see, whether such a whole-cell system or a combination of specialized (bio-) catalysts will eventually win the race to a commercial production of 86.

Cascade oxidation of 5-hydroxymethylfurfural 85 to 2,5-furan dicarboxylic acid 86 by multiple enzymes (dehydrogenases and/or oxidases) in vitro or in whole cells.133
Biocatalytic oxidation of cyclohexanol (87) can produce the polymer building block ϵ-caprolactone (88) and its oligomers (89) (Scheme 31).135 An initial study demonstrated the use of an alcohol dehydrogenase (ADH) and a Baeyer–Villiger monooxygenase (CHMO) to access 88, but with serious product inhibition at 60 mm.135a By using the unique acyltransferase activity of lipase CAL-A, 89 was produced at more than 20 g L−1 from 87 (200 mm).135c By using the hydrolysis activity of lipase CAL-B, also 6-hydroxyhexanoic acid was produced at a concentration of 283 mm (37.4 g L−1) from 87 in 20 h.135d Recently, the Bornscheuer group showed that a process with E. coli co-expressing ADH and CHMO and CAL-B produced 89 (>20 g L−1) in a 500-mL scale.135e The cascade has been extended to produce 6-aminohexanoic acid.135b ω-Hydroxy acids and lactones were also produced by oxidation of diols with ADH or alcohol oxidase.136

Cascade oxidation of cyclohexanol 87 to ϵ-caprolactone oligomers 89 by a combination of an alcohol dehydrogenase, a Baeyer–Villiger monooxygenase, and a lipase with acyltransferase activity avoiding the formation of undesired 6-hydroxy hexanoic acid.135
Besides oxidation from alcohols, acids can also be produced by biocatalytic oxidation of easily available hydrocarbons. The Park group developed the cascade oxidation of the C=C bond in oleic acid and linoleic acid into the diacids or ω-hydroxy acids, yet at a relatively low concentration.44, 137 The Li group reported the whole-cell cascade oxidation of styrene to (S)-mandelic acid, (S)- or (R)-phenylglycine, and phenylacetic acid in up to 140 mm (≈20 g L−1).138
One of the most challenging chemical reactions is selective oxidation of non-activated C−H bond (see Chapter 2.1). One of the most efficient examples is the oxidation of long-chain alkanes and fatty acids to α,ω-diacids by Candida tropicalis, a natural degrader of these compounds (Scheme 32).139 By deletion of four genes in the competing β-oxidation pathway, the C. tropicalis H5343 strain was engineered and employed for conversion of methyl myristate (90) to accumulate tetradecanedioic acid (92, 210 g L−1) in 162 h.139a In a more recent study, 16 genes involving oxidation of the ω-hydroxyacid 91 were deleted. With this engineered C. tropicalis DP428 strain, 174 g L−1 of 14-hydroxytetradecanoic acid (91) was accumulated by biotransformation of 90 (200 g L−1) within 148 h.139b The production of long-chain diacids by oxidation of alkanes with C. tropicalis is currently implemented on a commercial scale (40,000 ton year−1) in China by Cathay Biotech.23, 140 These examples illustrate the high efficiency of P450s for the conversion of the native substrates in the native host.

Cascade oxidation of methyl myristate 90 to the corresponding ω-hydroxyacid 91 or α,ω-diacid 92 by Candida tropicalis containing P450s, oxidases, and ADHs.139
4.3 Amides
Many small molecular pharmaceuticals contain amide bonds. Catalytic/direct amide formation with high atom economy has been identified as a key green chemistry research area by a panel of pharmaceutical companies.141 Biocatalysis could provide a possible solution for this challenge with a range of hydrolases (lipases, esterases, acylases) and ATP-dependent enzymes.142 The Penicillin G acylase had been well-established for the production of semi-synthetic penicillins and cephalosporins in industry.143 Furthermore, lipase-catalyzed amide-bond forming is the basis for the kinetic resolution of racemic amines (Chapter 3). While many lipases, amidases, and esterases are available, their application for amide formation is mainly limited to activated substrates (e.g. esters). Recently, a unique lipase was discovered for aminolysis of several esters (even acids) with several amines to produce amides in high yields in the presence of water (partially hydrated hexane).144
Amide synthesis with ATP-activation can directly use acids and amines in an aqueous environment. A team from GSK combined CoA ligases (CLs) and N-acyltransferases (NATs) to produce amides (Scheme 33).145 The preparative scale synthesis (<1 g) of an amide precursor (95) of Losmapimod was demonstrated with E. coli cells co-expressing 4-chlorobenzoate CoA ligase (CBL) and a serotonin hydroxycinnamoyl transferase (66CaAT). Glycerol was added to provide ATP through cellular metabolism. Acid 93 (10 mm) and an equimolar amount of amine 94 were successfully transformed to amide 95 in 83 % conversion and 74 % isolated yield.

Production of amide 95 from acid 93 and amine 94 using a combination of a CoA ligase (CBL) and a N-acyltransferase (66CaAT).
Another recent example for amide synthesis with ATP activation is from Grogan et al.146 An ATP-dependent amide bond synthetase (McbA) from Marinactinospora thermotolerans displayed a broad aryl acid specificity and gave access to a range of pharmaceutical-type amides. It was applied to synthesize Moclobemide (98) from the corresponding acid 96 (4 mm) and amine 97 (6 mm) with a facile ATP recycling system (Scheme 34).146b 98 was produced at 70 % conversion and 64 % isolated yield. Although these cascades are not efficient yet, enzyme engineering and optimized ATP recycling (Chapter 7.1) may enhance these concepts for environmentally friendly amide syntheses, particularly if the final driving energy is pulled from cheap inorganic polyphosphate.

Production of amide 98 from acid 96 and amine 97 using the ATP-dependent amide bond synthetase McbA and an ATP recycling system.
4.4 Sulfoxidations
A more recent addition to the biocatalysis portfolio has been the synthesis of chiral sulfoxides as synthons and precursors for API synthesis. Flavoproteins such as Baeyer-Villiger or styrene monooxygenases mainly provide access to these compounds. For the synthesis of Esomeprazole (API Nexium), the enantioselectivity of a Baeyer–Villiger monooxygenase was inverted and the enzyme was then improved for activity, stability and chemoselectivity by researchers from Codexis.147 Another example is the flavoprotein monooxygenase AbIMO from Acinetobacter baylyi ADP1, which was known to oxidize indole or styrene, but later it was found that it also accepts alkyl aryl sulfides and transforms them into the (S)-sulfoxides with high enantioselectivity (95->99 %ee). Using a whole-cell biocatalyst specific production rates of up to 370 U gCDW−1 were reported. This styrene monooxygenase was then used to make (S)-2-chloro-4-(methylsulfinylmethyl)pyridine at 30 g L−1 h−1 on the multi-kilogram scale.148
5 Glycosylation
Carbohydrates have the highest importance in the biosphere, not only because polysaccharides carry the largest part of organic biomass, but also because most other biopolymers (proteins and DNA/RNA) as well as many small biologically active compounds comprise sugar units. Latest since the Corona crisis the strive for antiviral drugs and vaccines has gained highest attention—and this is inevitably linked to carbohydrate chemistry since virus-host interaction is guided by glycans. However, carbohydrates are a particular challenge for the synthetic organic chemist, because the high number of marginally differentiated alcohol functions require sophisticated protecting group strategies to achieve selectivity in bond formation. The heavy use of protecting groups makes conventional organic synthesis inefficient. Since enzymes are inherently selective, they can bring tremendous efficiency gains to carbohydrate/glycan synthesis.
The very high activity of glycohydrolases has been commercially exploited for a long time: amylase, cellulases, and pectinases (to name just a few examples) have widespread use in polysaccharide hydrolysis and glucose production and have completely replaced acids as catalysts. A more synthetic use-case is the application of glucose isomerase for the production of fructose. With 14 million metric tons (dry weight) of high fructose corn syrup (HFCS; 42–55 % fructose, rest glucose and 1–4 % oligosaccharides) produced annually149 this is by far the largest (synthetic) biocatalytic process. The process is run continuously over immobilized glucose isomerase (IGI) at 60 °C. STY is approximately 1 kg L−1 h−1 (calc. on dry HFCS) and the catalyst consumption is low (ca. 0.05 g immob. enzyme kg−1 HFCS).150 The high activity of the isomerase allows work below the temperatures needed for base or Sn-catalysis, leading to a clean and selective reaction.151
Three types of carbohydrate active enzymes can be used synthetically to build anomeric bonds:
-
Leloir-type glycosyltransferases (“GTs”)—nature's tool to build up carbohydrates and glycosylate proteins. GTs need nucleoside diphosphate (NDP) activated glycosyldonors. This activation makes glycosylation energetically favorable and usually irreversible. GTs are highly selective without concomitant hydrolysis of products, but they are rather slow. Many of them are membrane-bound and often express poorly in E. coli. Hence, GTs have been accepted for lab scale preparation of complicated carbohydrates152 but have rarely found their way into kg-scale production.
-
Transglycosidases (“TGs”): These are glycoside hydrolases which have naturally or by mutation a reduced affinity for water as glycosyl acceptor. Thus, they can break glycosidic bonds and transfer the glycosyl donor to another sugar, establishing a new glycosidic bond. Since TGs are derived from glycosyl hydrolases, they are fast. However, hydrolysis is a frequent side-reaction. Withers has demonstrated a way to systematically avoid water activation and thus make hydrolases artificial “glycosynthases”.153 Since starting material and products of TG′s reaction often have similar glycosidic bonds, synthetic applications need to find ways to shift the equilibrium favorably.
-
Phosphorylases (“GPs”): Intermediate between the two classes above. Designed by nature to break down polysaccharides and maintain some of the energy of the anomeric bond. The glycosyldonor is not transferred to a water molecule (as in hydrolysis) but to phosphate. The reaction is reversible and a phosphorylase can use a glycosylphosphate as donor to make a new glycosidic bond.
A full and much more differentiated picture of carbohydrate active enzymes is given in the CAZy database (http://www.cazy.org).154
The number of preparatively useful enzymatic glycosylation has increased strongly over the last years. In the following paragraphs we can just give a few examples for illustration of the opportunities. The first example, Stevia, is meant to address the initial prejudice of GTs being too inefficient for large scale in vitro use.
5.1 Stevia Glucosides/ Rebaudiosides
Overweight and obesity has become more prevalent than hunger and poses enormous health problems. Hence, there is a need to reduce or replace caloric sweeteners like sucrose and HFCS from food and beverages. High intensity sweeteners like Aspartame can serve this need but lack full consumer acceptance as they are not perceived as natural. Stevia is a natural high intensity sweetener formed in high concentrations in the leaves of the plant Stevia rebaudiana. It is 200–300 times sweeter than sucrose and has thus a great substitution potential for sugar.

Natural steviol glucosides.
Stevia is a mixture of different steviol glucosides. Good cultivars of the Stevia plant deliver a mixture that is mainly composed of stevioside (Stev) 99 a and rebaudioside (Reb) A 99 b. The taste profile improves with the degree of glucosylation: while stevioside 99 a has an unpleasant after taste, Reb A 99 b taste is good. Best taste, almost as clean as sucrose, is delivered by Reb M 99 d. Unfortunately, the best tasting rebaudiosides Reb D 99 c and Reb M 99 d are only trace components in the plant extract.155 There is a race for the best way to Reb M on-going: the in vitro glucosylation of 99 a and 99 b to 99 c and 99 d was first demonstrated and patented by PureCircle/Coca Cola after the discovery of Reb M′s superior taste profile.156 The Stevia plant β-1,3-GT UGT76G1, expressed in E. coli, was the enzyme of choice, because it selectively glucosylates the 3-position only after introduction of a first β-1,2-branching glucose (avoiding a non-natural glucosylation pattern). With UGT91D2 the β-1,2-glucosylation of Reb A 99 b to Reb D 99 c was achieved, but with low and slow conversion (2 orders of magnitude slower than UGT76G1). PureCircle has cooperated with c-LEcta to engineer the GTs and claimed a transformation of Reb A 99 b to RebM 99 d scaled to the pilot plant and to the 25 g L−1 concentration level.157 The glucose donor is UDP-glucose (101) which is recharged with glucose from sucrose/sucrose synthase (SuSy; Chapter 5.2). CJ Cheiljedang patented the glucosylation of Stev 99 a to Reb A 99 b on the 100 g L−1 level, also using Stevia rebaudiana GTs expressed in E. coli.158
In an impressive piece of work, Codexis has engineered and optimized GT for a truly efficient process: all required enzymes (β-1,2-GT, β-1,3-GT and SuSy for glucosyldonor regeneration) have been engineered for higher activity, use of ADP-glucose instead of UDP-glucose as glucosyldonor, and heat stability at 60 °C.159 The heat stability and increase of reaction temperature from 40 to 60 °C delivered several process advantages: The E. coli background enzymes can be deactivated by heat shock, making enzyme purification obsolete. The solubility of substrates, intermediates, and product is increased, and the reaction is accelerated, delivering good STY. The starting material is a basic commercial material (“Reb A60”; composed of 60 % 99 b, 30 % 99 a, 10 % other steviol glucosides) which exhibits surprisingly high solubility compared to pure Reb A 99 b. After full conversion, the product Reb M 99 d crystallizes out of the reaction mixture (residual solubility:156a 1 g L−1), making separation and work-up easy. Enzyme consumption is quite low (ca. 5 g CFE kg−1 product, 0.5 g kg−1 ADP), giving up to 130 g L−1 product in 1 day (Table 6).160
Technology/ Enzyme |
Product |
Yield [%] |
Product conc. [g L−1] |
STY [g L−1 h−1] |
TTN (estim.) |
Catalyst load [g kg−1 product] |
Ref. |
|---|---|---|---|---|---|---|---|
Glucose isomerase |
Fructose in HFCS |
42 % conv., >99 % yield |
200 |
250 |
105 |
0.05 (immob. cat) |
|
GT |
Reb M |
>95 % |
130 (insoluble) |
5 |
– |
5 (CFE) |
|
TG (Glucansucrase) |
Reb A α-glucosides |
94 % (conv.) |
270 |
90 |
– |
ca. 60 |
|
GT (SuSy) |
UDP-glu |
86 % |
100 |
10 |
– |
10 (CDW) |
|
GP (LmSP) |
Gly-glu |
89 % |
224 |
10 (initial rate >300) |
– |
0.5 (calc. as pure enzyme) |
|
GP (BaSP) |
Kojibiose |
83 % |
570 |
8 |
– |
4 (CFE) |
|
GP (SuSy + CBP) |
Cellobiose |
70 % |
170 |
7 |
– |
– |
|
TG (Glucansucrase) |
Poly-α-1,3-glucan (mutan) |
20–80 % |
20 (insoluble) |
1 |
– |
2 (protein in CFE) |
|
TG (Hexoseaminidase) |
Lacto-N-triose II |
86 % |
280 |
3×103 |
– |
3 (CFE) |
The taste of Stev 99 a and Reb A 99 b can also be improved by α-1,4-glucosylation of the C19 sugar. Cyclodextrin glucotransferase (CGTase, a TG, not a GT!) transfers a string of 1–20 glucose units (mostly one to four) from cyclodextrin or from starch onto the stevioside backbone (“Hayashibara process”).161 The product is approved and introduced to some markets as “Glucosylated Stevia Leaf Extract”. CarbExplore utilizes a glucansucrase to glucosylate on the C19-sugar—here the new bond is α-1,6.162 This synthesis is highly efficient, reaching 270 g L−1 in 3 h with 94 % conversion.163 However, the α-glucosidic products are not truly natural. Grace to their high STY these combined plant extraction & in vitro glucosylation approaches may be competitive with the fully fermentative approaches in recombinant yeast, spear-headed by Evolva/Cargill164 and now best developed by DSM165 and Amyris.166
5.2 Nucleotide Activated Sugars
GT-catalyzed glycosylations require nucleotide activation of the donor-sugars. In the above illustrated β-glucosidation of Stevia, UDP-glucose (101) is the natural donor, but ADP-glucose or other nucleoside diphosphate glucose (NDP-glu) can also be used in vitro. Stoichiometric use of 101 would be too expensive for large scale application, despite the recently described efficient synthesis on the 100 g scale.167 Thus, it is common art to recycle the nucleoside diphosphate in situ by glucosylation of the NDP with sucrose (100) under catalysis of sucrose synthase (SuSy; Scheme 36; shown for UDP).168

Sucrose synthase (SuSy)-catalyzed recycling of nucleoside diphosphates such as UDP.168
Sucrose contains a very energy-rich anomeric bond. The Gibbs free energy of hydrolysis ΔGhyd of sucrose is −27 kJ mol−1, much higher than ΔGhyd of maltose (−15 kJ mol−1) or cellobiose (−12 kJ mol−1), even higher than ΔGhyd of glucose-α-1-phosphate (102). This is the basis for all sucrose driven glucosylations.169 It allows to have high concentration of 101 in the equilibrium (the lower the pH the more). The subsequent GT-catalyzed glucosylation of an acceptor other than fructose (e.g. the C19-glucose of Stev 99 a) with liberation of UDP is practically irreversible.
5.3 α-Glucosylation using Sucrose Phosphorylases
Sucrose phosphorylase (SP) catalyzes the reversible conversion of sucrose with inorganic phosphate to α-glucose-1-phosphate (102) and fructose (Scheme 37).

α-Glycosylation using sucrose phosphorylase (SP).
SP has an interesting promiscuity, allowing phosphate to be replaced by other acceptors while at the same time showing little tendency to hydrolyze substrate or products. This makes SP valuable for preparative glucosylations.170 The Nidetzky group took advantage of the promiscuity of the sucrose phosphorylase of Leuconostoc mesenteroides (LmSP) and transferred glucose from sucrose directly and selectively onto the secondary alcohol position in glycerol (Scheme 38). The transfer works under phosphate-free conditions; 102 does not appear as intermediate. 2-(α-Glucosyl)-glycerol (103) is formed in 90 % crude yield (63 % after chromatography) if glycerol is applied in excess (0.8 m sucrose; 2.0 m glycerol).171 The synthesis has been scaled to several hundred kg-scale (Table 6) by bitop AG that sells 103 as moisturizer for cosmetics.

Direct and selective glucose transfer from sucrose 100 to glycerol using a sucrose phosphorylase.171
The Desmet group has designed a process to make the rare sugar kojibiose (2-O-α-d-glucopyranosyl-d-glucopyranoside, 104) on the kg-scale with lab equipment.172 Mutants of the Bifidobacterium adolescentis sucrose phosphorylase (BaSP) selectively transfer the glucose unit of sucrose onto the 2-hydroxy position of a second molecule of glucose (Scheme 39). The liberated fructose molecule is transformed with glucose isomerase (GI) to glucose and serves as acceptor molecule, thus making the process highly atom efficient and elegant.

Synthesis of Kojibiose with a sucrose phosphorylase (BaSP) and a glucose isomerase (GI).172
Sucrose phosphorylase is just one of many glycoside phosphorylases. The combination of the sucrose phosphorylase (to make α-glucose-1-phosphate in situ) and the inverting cellobiose phosphorylase (CBP) has been employed already by Kitaoka in the 90′s to make cellobiose (4-O-β-glucopyranosyl-glucose), a disaccharide with a β-anomeric linkage.173 This enzymatic synthesis has now been developed to the 100 t/a scale by Pfeifer & Langen in cooperation with c-LEcta for diverse food applications.174 For a broader overview on the opportunities of phosphorylases the reader is referred to Field et al. and Desmet et al.175
5.4 Glucansucrase-Catalyzed Polymerization
Another way to utilize the high energy of sucrose is the polymerization achieved by a class of transglycosidases (TGs) called “sucrases”—either “glucansucrases”, if they polymerize the glucose unit of sucrose, or “fructansucrases”, if they polymerize the fructose unit. Different glucans are formed, depending on the bond-formation of the sucrase: for example, dextrans with α-1,6-linkages between the glucose units, or mutans 105 with α-1,3-linkages (Scheme 40). Glucansucrases are processively working enzymes. As TGs they are fast (kcat>100 s−1) and do not need co-factors or nucleotides. Very high molecular weights (Mw of 109 for a dextran) have been reported by the group of Remaud-Siméon.179 Glucansucrases and their application in nutrition have been pioneered and reviewed by the groups of Monsan180 and Dijkhuizen.181

Sucrase-catalyzed conversion of sucrose to the α-1,3-polyglucan Mutan 105: an enzyme catalyzed polymerization.
DuPont has investigated the use of low molecular weight glucans (mostly α-1,3-glucans) as low-glycemic soluble fiber in food.182 A mutanase was added to the sucrase-catalyzed polymerization to cut growing chains and keep polymers soluble. Unless heavily protected by α-1,2-glucosyl-branches these polyglucans remain more readily digestible than anticipated and led to the failure of the project. In recent patents DuPont describes glucansucrases that give a decent degree of α-1,2-glucosyl branches on the mutan backbone.183 Remaud-Siméon has also reported specialized sucrases to graft α-1,2-glucosyl-branches on polysaccharide backbones.184 Thus, the target to make nutritional valuable fiber may still be accessible.
DuPont has further engineered the synthesis and properties of the high molecular weight glucans and carried the glucan polymerization on the 20 m3 scale (Table 6). The obtained polyglucans went into application testing as composite fiber/filler for thermoplastics and rubber (to replace carbon black and silica in tires), coatings, paper coating, laundry, and adhesives.185 Yields of the insoluble polyglucan ranged between 20 % to 80 % of theory based on consumed sucrose, since considerable amounts of oligosaccharides were formed. A major challenge in up-scaling is probably the separation of the hydrated but insoluble, non-crystalline, low density polymer from the fructose solution by filtration or centrifugation.
Interestingly, even the α-1,4-glycosidic bond carries still enough free energy to enable molecular weight increase, if relaxed to an α-1,6-glycosidic bond: Ezaki Glico has developed a process to convert starch derived low molecular weight amylose (only α-1,4-links; Mw ca 104 Da) to higher molecular weight glycogen mimics composed of α-1,4-linked polyglucose with a high density of α-1,6-branches (Mw range 3 to 30×106 Da).186 The key enzyme is a TG, the amylase related “branching enzyme” from A. aeolicus supported by an amylomaltase. This process offers access to plant based glycogen for food purposes (e.g., athlete's nutrition).
5.5 Perspectives of Biocatalytic Carbohydrate Production
There is a rapid increase of examples of successful biocatalytic syntheses in the carbohydrate arena which will give the rising research field of carbohydrates an important impulse. As it can be concluded from the KPIs of the described processes (Table 6), biocatalysis on carbohydrates can work at very high concentrations. In fact, many carbohydrate active enzymes are rather stabilized by high sugar concentrations, in clear contrast to enzymes that are challenged with less polar, second phase forming organic substrates or solvents. Thus, biocatalysis will become an increasingly important tool to provide the larger amounts of carbohydrates necessary for clinical studies on health benefits and eventually commercial production. An up-coming topic in this field will be the human milk oligosaccharides which currently profit mostly from fermentation technology but may require additional biocatalytic glycosylation for second and third generation products.178, 187
6 Complex Molecules
Biocatalysis has traditionally been used for functional group interconversion as highlighted by the examples in the previous chapters. Within the last decade however, the biocatalysis community has begun to actively investigate the development of molecular complexity and scaffold synthesis that traditionally has been the purview of the organic chemist. Of particular focus has been the carbon-carbon bond-forming reactions (reviewed previously97), many of which introduce only small changes in molecular weight and complexity. However, a few have demonstrated the potential to suggest new routes to stitching molecules together. These include the carbon-carbon bond forming enzymes of the lyase family, such as norcoclaurine synthase (Chapter 6.1.1, officially classified as a carbon-oxygen lyase), the aldolases described in Schemes 21 and Chapter 6.1.3) and the tryptophan synthases highlighted in Chapter 6.1.2 as examples. Molecular complexity is also developed in carbon-nitrogen bond forming enzymes like the reductive aminase IREDs (Scheme 14 and Vernakalant79 as examples), the amide forming peptide synthases and carboxylic acid reductases (Schemes 34 and 45, respectively), and the nucleoside phosphorylases for nucleosides exemplified above. Some of these chemistries also take advantage of high energy phosphate bonds in order to drive reactions forward (Schemes 34, 43 and 45) highlighting an increasing interest in kinases, phosphorylases and ATP recycling systems.
6.1 Reactions Developing Molecular Complexity
6.1.1 Norcoclaurine Synthase
Norcoclaurine synthase (NCS) is a single enzyme that in nature brings together dopamine and 4-hydroxyphenylacetaldehyde to form (S)-norcoclaurine (Scheme 41 a). Mechanistically, the enzyme forms an imine between the amine and aldehyde, then creates a C−C bond between an aromatic carbon and the carbon of an imine using a Pictet-Spengler reaction mechanism, leading to the benzylated tetrahydro-isoquinoline privileged scaffold.188 The enzyme has demonstrated a broad substrate scope for aldehyde substrates (Schemes 41 b,c), and under non-optimized laboratory conditions delivered non-canonical tetrahydroisoquinoline products with a remarkable STY of 7 g L−1 h−1 (Scheme 41 b).189, 190 In addition, the enzyme has responded to mutation with improved performance, highlighting its evolvability toward increasingly-complex reaction partners.190 A final example demonstrates this enzyme's ability to function in cascade reactions, further highlighting its utility.191

6.1.2 Tryptophan Synthase
Tryptophan synthase synthesizes tryptophan from the substrates serine and indole as a second example of a lyase family member. Mechanistically, the enzyme forms an imine between the amine in l-serine and the aldehyde of the PLP cofactor, allowing through a series of concerted aldimine-ketimine intermediates, the removal of the serine alcohol to form a terminal alkene in its place, followed by subsequent attack of the alkene by the indole. The reaction was initially valuable for its ability to execute this chemistry on a variety of substituted indoles that are synthetically challenging to make.192
Recent advances call attention to the unique molecular complexity that this enzyme can provide. Variants developed added methylserine as an accepted substrate, leading to the formation of β-methyl tryptophan derivatives (Scheme 42 a)193 and 3-substituted oxindoles, leading to quaternary centers on the indole at the point of attachment (Scheme 42 b).194

6.1.3 Aldolase and Nucleoside Phosphorylase
Enzymes are well suited to cascade reactions, because they are naturally designed to work in similar environments of solvent, temperature and other environmental conditions, and because they come with an inherent selectivity that allows several catalysts to work in concert with little fear of cross-reactivity. In cases where the cascades are designed to develop molecular complexity, complex molecules can be built with remarkable efficiency. An additional collection of enzymes in the lyase family that creates difficulty to achieve complexity is the aldolases. The degree of complexity introduced by the aldolases is low when examined by molecular weight of the corresponding products, but they are responsible for the synthesis of a wide variety of sugars, which makes them valuable. By combining the sugar made with a second complexity generating step, a C−N forming enzyme from the nucleoside phosphorylase family, a complex nucleoside could be readily made. In fact, this is the synthesis design for the manufacturing route to Islatravir 123 (Scheme 43) for the treatment and pre-exposure prophylaxis of HIV.195

An enzyme cascade with nine enzymes to produce Islatravir from simple starting materials.195
This synthesis is a three-step, one-pot cascade starting from alkynyl glycerol, with the molecular complexity formation coming from deoxyribose phosphate aldolase (DERA) and purine nucleoside phosphorylase (PNP) in the third and final step. The first reaction sets up the aldolase by producing the necessary aldehyde and simultaneously desymmetrizing alkynyl glycerol. The second reaction utilizes a kinase to introduce the phosphate that will eventually serve as the leaving group during installation of the fluoroadenine base. Note that this reaction uses catalytic ATP that is recycled with acetyl-phosphate and acetate kinase, different from ATP recycling previously described (Scheme 34). The third and final reaction contains all of the complexity building steps: the DERA begins by combining the alkynyl aldehyde and acetaldehyde added at this step to form the 5′-phosphate alkynyldeoxyribose. After phosphopentomutase (PPM) moves the 5′-phosphate to the 1′-position, the second complexity building step is executed when PNP catalyzes the displacement of the 1′-phosphate with fluoroadenine. The cascade produces the active pharmaceutical ingredient in a single reaction vessel from simple, low molecular weight building blocks and fluoroadenine in 51 % overall yield, and represents the state of the art in chemical synthesis for this molecule. This last step has the lowest volumetric productivity of the three, with a STY of 16.6 g L−1 d−1.
6.2 Ex vivo Natural Product Biocatalysis
The high complexity achieved in the biosynthesis of metabolically engineered whole-cell systems have inspired biochemists to re-establish anabolic enzyme cascades in vitro/ ex vivo and to avoid the side-reactions of the full metabolism of a living cell. While not yet ready for large scale production, recent progress is remarkable (STY and product titers in some cases already exceeding fermentation yields) and the space between single step biocatalysis (in vitro) and fermentation (in vivo) is being filled.196 The advantages and disadvantages of cell free cascades have been critically discussed recently.197
A very recent development in the biocatalytic arsenal is the ability to overexpress and isolate the enzymes in natural polyketide synthesis pathways and execute a natural product synthesis in a chemical facility. While only a handful of examples exist (for the earliest publications see ref. 198), and these examples are not commercialized and therefore not scaled, they do represent an amazing feat of complexity-building biocatalysis. A recent example for this success is the Ikarugamycin (126) synthesis199 (Scheme 44).

An ex vivo enzyme cascade to produce Ikarugamycin 126 from simple starting materials.199
In this example, three competing chemical routes to Ikarugamycin starting from commercially available materials produced Ikarugamycin in 27, 29 and 32 steps. In contrast, the three His-tagged and affinity-purified natural enzymes when mixed together with acetyl-CoA, malonyl-CoA, and the amino acid l-ornithine produced Ikarugamycin in a single overnight reaction in 9 % isolated yield. Adding three additional enzymes and the substrates acetic acid and malonic acid allowed the recycling of catalytic amounts of CoA cofactor, still in a single reaction. Although the reaction productivity is low (STY 0.08 g L−1 d−1) and the reaction has not been scaled (largest reaction reported in 3.7 mL volume), the reaction has also not undergone process intensification, and the enzymes have not been evolved to optimize their performance ex vivo. In this case, the polyketide synthase (ikaA) was responsible for the complexity as defined by molecular weight increase, while the second enzyme (ikaB), likely a desaturase, is responsible for the initial formation of the broader scaffold.
7 Biocatalytic Reactions for Potential Industrialization
Over the last decade, a range of novel enzymes have been (re)-discovered for future applications in organic synthesis. Many of them have been known for decades and they often had been biochemically characterized, but their use in biocatalysis only became prominent recently. Reasons have been that either their potential had been overlooked, new reactions types catalyzed by them were just discovered, intelligent solutions to overcome practical limitations have been developed, and most importantly, or methodology to discover and develop good quality enzymes (such as novel tools for enzyme discovery in sequence databases, better expression systems to make them, enzyme engineering to improve their performance, selectivity and substrate scope) have been developed.
A very recent example has been the development of an efficient enzymatic process for the recycling of PET (polyethylene terephthalate), in which pretreatment of PET from waste bottles, substantial enzyme engineering and bioprocess development enabled to isolate the monomer terephthalic acid (TA) after 90 % depolymerization at a mean productivity of ≈17 g L−1 h−1. New PET made from this monomer matches the quality of virgin PET and the overall process now provides the basis for a higher circularity for the widely used polyester PET despite the additional cost associated with the enzyme and base demanding depolymerization.200 Furthermore, thanks to advanced methods for enzyme engineering, computational enzyme design and research across catalytic disciplines, a range of novel often called “new-to-nature” chemistry reactions have been implemented into biocatalysts.
This section will therefore briefly exemplify where the biocatalysts are to our knowledge not yet used on large (industrial) scale, but the potential is present.
7.1 Carboxylic Acid Reductases
Carboxylic acid reductases (CARs) are useful biocatalysts to make aldehydes from carboxylic acids under mild reaction conditions. These reactions are especially of interest for the flavor and fragrance industry where aldehydes are important products, but also for various applications in chemistry as summarized in recent reviews, which also cover in detail the current status on the discovery, structures and applications of CARs.201 Compared to chemical catalysts to reduce a carboxylic acid to the aldehyde, the commonly observed undesired over reduction to the alcohol does not take place with CAR (unless in a whole-cell system where other reductases from the host are present). Expression of active CARs requires a suitable phosphopantetheinyl transferase (PPTase), and for the reduction reaction they also require the cofactors NADPH and ATP in stoichiometric amounts. In addition to the discovery of various CARs and PPTases from various microorganisms, one breakthrough was the recent elucidation of the first structures of the CARs from Nocardia iowensis and Segniliparus rugosus.202 For this, a high-throughput assay has been described and successfully applied to identify better CAR variants in mutant libraries.203 For their application, whole-cell systems appear suitable as the ATP can be readily recycled, but the aldehydes formed are usually toxic to the host such as E. coli. This was overcome by Bayer et al. by coexpression of an alcohol dehydrogenase and a CAR to ensure that only a tolerated aldehyde concentration was present in the host cell while a co-expressed dihydroxyacetone-dependent aldolase then readily converted the aldehyde formed with DHA to yield the desired aldolase product in 70 % overall yield.204 Alternatively, Turner and co-workers have shown that CARs can be used as isolated enzymes in a reaction cascade.205 This allows a combination with two further enzymes to afford substituted piperidines and pyrrolidines.
Thus, the aldehyde formed by the CAR was converted by an amine transaminase (ATA) to the ω-amine α-carboxylic acid that underwent in situ ring closure to the imine followed by stereoselective reduction catalyzed by an imine reductase (IRED) to the product at excellent overall conversion and optical purities (Scheme 45).205 Notably, CARs have also been used for the reduction of dicarboxylic acids to afford the corresponding diols such as 1,4-butanediol or 1,6-hexanediol, useful precursors to make polymers.206

An enzyme cascade comprising a carboxylic acid reductase (CAR), a transaminase (ATA) and an imine reductase (IRED) enables production of piperidines and pyrrolidines in high yield and optical purity.205 (n=0, 1).
Interestingly, Flitsch et al. demonstrated that CARs can also be used to convert carboxylic acids into the corresponding carboxylic amides, where the adenylation domain of the CAR in the presence of ATP and nucleophiles such as amines, directly yields the target tertiary amide, notably only in the absence of NADPH to avoid aldehyde formation.207 Very important for larger scale applications of CARs, it could be shown that ATP recycling from AMP can be performed efficiently in vitro using pyrophosphate (PPi) in combination with a polyphosphate kinase,208 a system initially developed for SAM-dependent methyltransferases.209 As pointed out in a recent review,201b at present the productivities of CAR-catalyzed reactions and the scale on which they have been performed are some way off being viable for truly large scale industrial synthesis. Nevertheless, these obstacles also applied to many other enzyme classes until some years ago and efforts in enzyme discovery, engineering and bioprocess design allowed to overcome these limitations.1c
7.2 Methylation and Demethylation Reactions
Regioselective methylation as well as demethylation play an important role in natural product and metabolite modification as well as in the synthesis of pharmaceuticals. Naturally, methylation is commonly catalyzed by S-adenosyl methionine (SAM)-dependent methyltransferases (MT) which can transfer the methyl group from this cofactor to oxygen, carbon, sulfur, nitrogen, or phosphorous. Until recently, the biocatalytic application of MTs, of which many could be expressed as recombinant enzymes and also engineered for new target reactions,211 has been hampered by the stoichiometric requirement of SAM (132). One option is the use of pyrophosphate with a polyphosphate kinase as described above in section 7.1, but this requires in total addition of six enzymes,209, 212 making it rather complex. Another recently published alternative is the use of a halide methyltransferase (HMT) where methyl iodide is used as donor, and for five different MTs up to 290 cycles were reported (Scheme 46).210 It should be noted, however, that methyl iodide is a rather toxic and expensive reagent.

A halide methyltransferase (HMT) converts S-adenosyl homocysteine (SAH, 131) with methyl iodide into S-adenosyl methionine (SAM, 132) serving as methyl donor for methyl transferases.210
Similar to methylation, the regioselective demethylation, especially under mild conditions, is an important biocatalytic reaction. This reaction can be catalyzed by P450-monooxygenases via hydroxylation of the methyl group using molecular oxygen and NADPH, where the resulting unstable product undergoes decomposition into the free hydroxyl group and formaldehyde. The Flitsch group described a range of robust and practically versatile self-sufficient P450 enzymes, which were able to demethylate a range of substrates (Scheme 47).213 The Bornscheuer group discovered P450s in marine bacteria, which selectively demethylate 6-O-methyl-d-galactose 135 present in algal polysaccharides, such as agar and porphyran. These enzymes need redox partner proteins for their activity, and their biochemical function as well as their structural features have been elucidated.214

Demethylation was shown using several self-sufficient P450-monooxygenases for a range of methyl-product aryl ketones215 as well as for the demethylation of 6-O-methyl d-galactose using an enzyme from a marine bacteria, which requires redox partner proteins (FoR, FoX).214 These reactions yield formaldehyde as by-product.
7.3 Halogenation Reactions
Enzymatic halogenation has the potential to develop greener and more selective processes for the production of important halogenated compounds as the use of halogenases in aqueous reaction media at ambient temperatures with distinct chemo- and regioselectivity has substantial advantages over chemical halogenation methods. Although the first halogenating enzymes were discovered a long time ago, the past few years have seen substantial progress in the discovery of novel enzymes as well as their improvement by enzyme-engineering including their application in preparative scale synthesis. Biohalogenation has been reported for a broad range of enzymes including heme-iron or vanadium-dependent haloperoxidases, α-ketoglutarate-dependent halogenases, flavin-dependent halogenases as well as monooxygenase, fluorinases216 and very recently iodanases. Excellent recent reviews217 cover this area in detail and in the following hence only selected examples are covered.
Stimulated by the discovery and structure elucidation of the l-Trp 7-halogenase (PrnA), which regioselectively chlorinates tryptophan, by the van Pée group,218 numerous scientists studied since then these biocatalysts with focus on alteration of halide preference, regioselectivity, activity and stability. The best studied enzymes are RebH, ThaI, PrnA and PyrH, which were subjected to substrate profiling for a range of anilines, indoles, pyrroles and azoles.217b, 219
The Sewald group demonstrated gram scale synthesis of 7-Br-l-Trp using RebH in combination with the flavin reductase PrnF224 and cofactor recycling of NADH using an alcohol dehydrogenase as immobilized “Combi-CLEC” catalysts.222a More recently, the same group substantially improved the thermostability of the ThaI halogenase by directed evolution and semi-rational design to yield 6-Br-l-Trp.222b Also a family of radical halogenases were described, which produce mono- and dichlorinated, as well as brominated and azidated, amino acids.225
Recently the chemo-, regio- and diastereoselective chlorination of an unactivated C(sp3)-H bond was achieved after directed evolution of the FeII and α-ketoglutarate-dependent halogenase from Westiella intricate (WelO5, Scheme 48).220 In another study, the related enzyme WelO5* as well as the halogenase AmbO5 were evolved and showed distinct patterns of regioselectivity (Scheme 48).221

Halogenation is not restricted to chloride or bromide and recently the first iodinase was described.223 By genome mining, a viral halogenase (VirX1) was discovered that catalyzes the halogenation of a range of arenes as exemplified for 32 different heterocyclic compounds. This enzyme shows substantially higher activity for iodide over bromide and very poor chlorination.226 Thus, halogenase enzymes are on the way to become useful biocatalysts, especially for late-stage functionalization of pharmaceuticals.
7.4 Aldoxime Dehydratases
Nitriles are produced chemically on large industrial scale especially for bulk chemicals such as acetonitrile or acrylonitrile, but also are important intermediates for pharmaceuticals. However, synthetic routes to nitriles require the use of highly toxic cyanide. An attractive alternative is are aldoxime dehydratases, first described for synthetic use by Kato and Asano.227 These enzymes can convert an aldoxime in one step through dehydration directly into nitriles and have a surprisingly large substrate scope: aliphatic and aromatic aldoximes are converted into the corresponding nitriles.228 The enzymes are robust and can dehydrate essentially pure substrate without the need for dilution. BASF has developed a synthesis of fragrance nitriles (example: citronellylnitrile 142, Scheme 49) by treating crude citronellyloxime 141 undiluted with Bacillus sp. phenylacetoxime dehydratase (PAOx) overexpressed in E. coli.229 A high STY (19 g L−1 h−1 until 50 % conversion, 8 g L−1 h−1 until total conversion) and a final titer of 0.7 kg L−1 were observed. Gröger et al. described very high titers when converting neat octanaloxime to octylnitrile as well as high enantioselectivities (Scheme 50).230

Synthesis of the fragrance citronellynitrile using a phenylacetoxime dehydratase (PAOx).229

An aldoxime dehydratase (OxdA, used as whole cell system) catalyzes the stereoselective formation of a nitrile from the aldoxime.231
Interestingly, as the enantiopreference depends on the E and Z isomers of the aldoxime, opposite enantiomers can be obtained from the same racemic aldehyde.231 The combination of hydroformylation and biocatalysis has been shown.232
7.5 “New-to-Nature” Chemistry
In the past decade, a range of novel biocatalytic applications have been developed, especially via enzyme engineering, where reactions have been made possible, for which no counterpart is known from nature. These examples have in common that larger scale applications have not been shown yet, but the potential is there. Readers are referred to a range of reviews covering also artificial metalloenzymes and biocatalysts based on non-natural genetically encoded amino acids.233
One example is the formation of cyclopropanes (146) from styrene using an engineered P450 monooxygenase (P411 variants derived from the P450 enzyme BM3 from Bacillus megaterium). These P411 biocatalysts can react with the diazo reagent ethyl diazoacetate to catalyze a carbene transfer instead of natural oxygen incorporation (Scheme 51).234 This was also shown to work with high diastereo- and enantioselectivity using engineered myoglobin.235 Similarly, aziridines236 could be accessed from tosyl azide as precursor, but the TTNs are rather low.

Examples for “new-to-nature” chemistry catalyzed by engineered biocatalysts.
The Hyster group pioneered the asymmetric dehalogenation of α-bromo-α-aryl/alkyl lactones such as 147 using an NADPH-dependent KRED as protein scaffold, which upon light irradiation afforded the dehalogenated chiral lactone product 148 (Scheme 51).237 This concept was then expanded to the photoexcitation of flavin-dependent ene reductases (ERED).238 With an artificial metalloenzyme the olefin metathesis using Grubbs catalyst within a streptavidin scaffold, expressed in the periplasm of E. coli, was possible but not efficient (Scheme 51).239
8 Conclusion and Outlook
An impressive range of industrial scale biocatalytic applications have been developed since the previous reviews2 were published. When looking at recent examples, a remarkable achievement emerges—novel enzymes, which were discovered in academic laboratories only 5–7 years ago, have already been engineered and applied on scale. This shortened development time, expanded enzymatic portfolio and advanced applications, for example, demonstration of an enzymatic cascade for preparation of an API, will contribute to the continued adoption of biocatalysis in synthetic chemistry, overcoming the inertia we have seen in the previous decade.
Process engineers may be pleased to see that many biocatalytic reactions now achieve STY comparable to conventional chemical transformations. Most enzymes are highly efficient—it takes very little enzyme to make a lot of chemistry. Thus, it only takes small pilot-plant equipment (and very standard expression technology) to supply enough catalyst for regular production. More and more biocatalysts are available commercially on kg-scale. High selectivities and clean reactions facilitate down-stream-processing. This makes biocatalysis easier to adopt in chemical production than fermentation, as fermentation has a 1–2 orders of magnitude lower STY—and hence a need for huge fermenter volumes and sophisticated metabolic engineering to create the producing organism. Effectively, the problem statements given in Table 1 have been overcome.
Looking ahead, further advances in enzyme and reaction engineering, machine learning, dynamic enzyme reaction modelling, and predictive retrosynthetic tools, will embed enzymatic synthesis as a green, sustainable, cost- and atom-efficient method for the manufacture of ever-increasing molecular complexity across the chemical, pharma, and food industries.
Acknowledgements
S.W. thanks the Alexander von Humboldt foundation for a Humboldt research fellowship. Open access funding enabled and organized by Projekt DEAL.
Conflict of interest
R.S. is an employee of Novartis, J.C.M. is an employee of Merck & Co, Inc. All have been involved in inventions cited in this review. K.B. declares to own stock of several of the companies mentioned in the review.
Biographical Information
Shuke Wu obtained his PhD from the National University of Singapore (2015) with Prof. Zhi Li (NUS) and Prof. Daniel I. C. Wang (MIT). In 2017, he moved to Switzerland to work with Prof. Thomas R. Ward at the University of Basel sponsored by a Swiss government excellence scholarship. Currently, he is working with Prof. Uwe T. Bornscheuer at the University of Greifswald as an Alexander von Humboldt fellow.

Biographical Information
Radka Snajdrova obtained her PhD from Vienna University of Technology in 2007, followed by postdoctoral studies at York University (UK) and Greifswald University (Germany). In 2010, she transitioned to industry applying and developing biocatalytic technologies at Novacta in the UK, prior to joining Chemical Process Development at GSK, with responsibility for the development and implementation of new biocatalytic technology in both pre- and post-commercialization routes. Since Dec. 2016, Radka is leading the Bioreactions group in GDC at the Novartis Institute for Biomedical Research in Basel, Switzerland.

Biographical Information
Jeffrey Moore obtained his PhD in Chemical Engineering from the California Institute of Technology in 1996 as Frances Arnold's first Directed Evolution graduate student. His foundational work led to an evolved p-nitrobenzyl esterase and the Lonza Centenary Prize (1997). In 1996, he joined the Biocatalysis Group of Merck & Co.in Rahway NJ, spending two decades inventing new enzymes and new enzymatic processes. In 2018, he transitioned to the Merck Protein Engineering Group responsible for evolving enzymes for the discovery, development and commercial scale manufacture of medicines. He has been awarded a US Presidential Green Chemistry Award (2010), the BioCat2012 Award (2012) and the Thomas Edison Inventorship Award (2014).

Biographical Information
Kai Baldenius studied chemistry in Hamburg and Southampton. He received his PhD for research in asymmetric organometallic catalysis, supervised by H. tom Dieck and H. B. Kagan. After his postdoc on natural product synthesis with K. C. Nicolaou at the Scripps Research Institute he joined BASF in 1993. Kai served BASF in various functions (R&D, production, marketing, sales) before he took the lead of BASF′s biocatalysis research for almost a defcade. He left BASF to become a free-lancing consultant in 2019 and in 2020 he has founded Baldenius Biotech Consulting.

Biographical Information
Uwe T. Bornscheuer studied chemistry and received his PhD in 1993 at Hannover University followed by a postdoc at Nagoya University (Japan). In 1998, he completed his Habilitation at Stuttgart University about the use of lipases and esterases in organic synthesis. He has been Professor at the Institute of Biochemistry at Greifswald University since 1999. Beside other awards, he received in 2008 the BioCat2008 Award. He was just recognized as “Chemistry Europe Fellow”. His current research interest focuses on the discovery and engineering of enzymes from various classes for applications in organic synthesis, lipid modification, degradation of plastics or complex marine polysaccharides.


