

Mechanistic Studies in Photocatalysis
Graphical Abstract
Lighting the (reaction) path: Photoredox and photocatalysis have recently provided fresh opportunities to expand the potential of organic synthesis. So far, innovation has mainly been driven by the quest for novel reactivities, often at the expense of a thorough mechanistic understanding. But these fields are now entering a more mature phase where the combination of experimental and mechanistic studies will play a dominant role in sustaining further innovation.

Abstract
The fast-moving fields of photoredox and photocatalysis have recently provided fresh opportunities to expand the potential of synthetic organic chemistry. Advances in light-mediated processes have mainly been guided so far by empirical findings and the quest for reaction invention. The general perception, however, is that photocatalysis is entering a more mature phase where the combination of experimental and mechanistic studies will play a dominant role in sustaining further innovation. This Review outlines the key mechanistic studies to consider when developing a photochemical process, and the best techniques available for acquiring relevant information. The discussion will use selected case studies to highlight how mechanistic investigations can be instrumental in guiding the invention and development of synthetically useful photocatalytic transformations.
1 Introduction
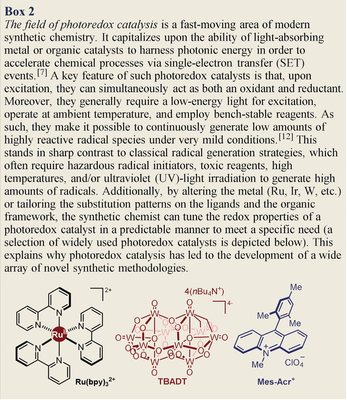
Synthetic photochemistry deals with electronically excited molecules and the chemical processes induced by light (Box 1).1 Since the chemical reactivity of excited molecules differs fundamentally from that in the ground state,2 light-mediated chemistry has the potential to unlock reaction manifolds that are unavailable to conventional ground-state pathways. Chemists recognized this potential in the early days of organic synthesis.3 However, synthetic photochemistry remained a specialized area with limited practical applications. This situation changed dramatically 10 years ago, when seminal independent works from MacMillan,4 Yoon,5 and Stephenson6 conceptualized the field of photoredox catalysis.7 This approach exploits the ability of a visible-light-absorbing photoredox catalyst, upon excitation, to either remove an electron from or donate an electron to simple organic substrates (Box 2). This single-electron transfer (SET) mechanism facilitates access to highly reactive radical species under mild conditions. The synthetic community quickly recognized the new opportunities created by photoredox catalysis, and photocatalysis8 in general. Nowadays, thousands of researchers from academia and industry9 are developing and using light-driven processes to efficiently make molecules. As a result, niche fields such as radical chemistry10 and photochemistry (which could previously be mastered by only a few specialized chemists) have become widely used tools of modern synthetic chemistry. Over the last decade, great effort has been dedicated to developing new powerful methodologies. But this was at the expense of a more rational and mechanistically guided approach. The general perception today is that photocatalysis is entering a more mature phase, where the combination of experimental and mechanistic studies will play a dominant role in sustaining further innovation.
One may wonder why progress in photocatalysis has often relied on empirical findings rather than on a thorough mechanistic understanding of the photochemical process. One reason is that many new practitioners of light-driven chemistry come from the more traditional fields of polar chemistry and two-electron reactivity. The lack of familiarity with the classical experimental techniques most relevant to photophysical investigations and open-shell reactivity has hampered their extensive and proper application. In addition, these techniques were originally designed for theoretical purposes and not necessarily applied to guide the development of a methodology. Classical photophysical investigations to determine the excited-state kinetics and the underlying mechanism of a photochemical process are generally conducted under idealized conditions. In contrast, optimizing a synthetic method requires the systematic variation of many variables (e.g. temperatures, use of additives, high concentrations, excess of substrates), which means that the conditions are far from ideal. Applying photophysical techniques to such complex organic reaction matrices can be difficult, which complicates mechanistic determination under synthetically relevant conditions. In addition, the adjustment of these analyses to make them more applicable and accessible to synthetic laboratories can affect their consistency and precision.

Recently, renowned experts in radical chemistry13 and photo-physical studies14 have remarked upon the need to perform more accurate and thorough mechanistic investigations when developing a new methodology. These suggestions have already been partially embraced by the photocatalysis community, who have started to recognize the benefit of combining experimental and mechanistic investigations. While more efforts are needed along these lines, it is undeniable that the primary focus of a synthetic photochemist is (and should remain) the identification of novel reactivities to ultimately develop synthetically useful protocols. It would be beneficial if the (presently rather antagonistic) relation between synthetic and (photo)physical organic chemists could serve to foster collaborations. Joint efforts would help both groups appreciate and understand different perspectives and would undoubtedly be positive for the field of photocatalysis.
In this Review, we outline the key mechanistic aspects that should be carefully considered when developing a new synthetic photochemical process, and the best techniques available for acquiring relevant information. Ideally, we would like this text to serve as a roadmap to identify, following a temporal line, the proper mechanistic experiments to perform at different stages of methodology development: from the design of a novel reaction to the mechanistically informed development of the method. We will use selected case studies to highlight how mechanistic investigations can guide the invention and development of synthetically useful photocatalytic transformations. Here, our intent is not didactic. Readers willing to gain deeper insights into the theoretical aspects underlying classical photophysical investigations should refer to comprehensive and specialized review articles and books.15
2 Importance of the Experimental Set-Up
The reproducibility of an experiment is at the basis of the scientific method and the foundation upon which science advances.16 This applies to photochemistry too. When performing a photochemical reaction, chemists deal with a peculiar reagent: light. Many parameters can significantly alter the efficiency of a light-triggered process, including light intensity, the light source, and its distance from the reaction vessel.17 Recently, the photocatalysis community has recognized the need to enhance reproducibility to meet the standards of reliability demanded in both academic and industrial settings. An increasing amount of attention has been devoted to identifying more reliable experimental set-ups to standardize photochemical processes.18 Therefore, when assembling a set-up to run a photochemical reaction, it is essential to carefully consider and control specific parameters (Figure 1).

General experimental set-up and crucial parameters to control when performing a photochemical reaction.
Light source. The first law of photochemistry (the Grotthuss–Draper law) states that light has to be absorbed to cause a photochemical reaction.19 When choosing the irradiation system, the goal is to achieve selective and efficient irradiation of the chromophore, whether this is the substrate (direct photochemistry) or the photocatalyst (photocatalysis). This requires the chemist to maximize the overlap between the emission profile of the light source and the absorption spectrum of the absorbing species, while minimizing the excitation of other reaction components, which could lead to undesired reactivity. The choice of the best wavelength (λ) is therefore informed by the optical properties of the photoactive species, which can be simply evaluated by absorption spectroscopy (see Section 3.1).
Using sunlight to drive chemical processes is a fascinating possibility,3 and examples of efficient solar photochemistry have been reported.20 However, the use of solar light inherently undermines the reproducibility of a process because of the limited daily hours, weather dependency, and not uniform irradiation. Of the artificial light sources, compact fluorescent light (CFL) bulbs are a valid choice for explorative endeavors. Although CFL lamps provide moderate light intensity, they enable irradiation over a broad range of wavelengths (from ca. 250 to 720 nm), including UV spectral regions. Conversely, high-power light-emitting diodes (LEDs) are considered the preferred light sources for synthetic photochemistry. This is because they provide a narrow emission band (±20 nm), which is crucial to achieving selective irradiation of the chromophore and preventing unwanted side reactions. LEDs are generally cheap and sold in a broad selection of wavelengths (from ca. 250 to over 800 nm), which makes them ideal for reaction development. Excimer lamps are other quasi-monochromatic light sources of wide application in UV photochemistry.21 High-pressure Hg- and Xe-lamps, equipped with monochromators, because of their high intensity of emission, are generally used for precise analytical studies rather than for synthetic purposes (for example for quantum yield measurement, see Section 7). The same reasoning applies to lasers, which provide a powerful, monochromatic, and reliable emission. However, these features come at the expense of high costs and low versatility, thus thwarting their use for synthetic methods.
Irradiance. Another fundamental parameter to carefully control is the light intensity, or the applied irradiance (mW cm−2). The general idea is that an increased photon flux, by eliciting a proportionally higher photon-capture event, should linearly correlate with the rate of a photochemical process. But this is true only when the process is under a photon-limited regime. When optimizing a reaction, the target is therefore to apply the lowest intensity of emission that ensures light saturation. This scenario can be easily achieved when dealing with substoichiometric amounts of a chromophore. For example, when using a photoredox catalyst, which is present in low concentrations, light saturation is reached with a relatively low irradiance. The situation can dramatically change when the direct excitation of substrates is responsible for triggering the reactivity (direct photochemistry in Box 1). It is worth considering that irradiation at an excessive intensity may lead to the uncontrolled generation of highly reactive intermediates (which may trigger unwanted side reactions) or to substrate/product degradation. The use of an external power supplier connected to the light source readily enables the fine modulation of the irradiance, thus ensuring reproducible and optimal reaction conditions.
Temperature. Excited-state chemistry is generally independent of temperature effects. However, many photoredox catalytic processes rely on the generation of radicals, which are successively intercepted by reactive intermediates, including organocatalytic species22 or transition metal complexes.23 These are classical intermediates for thermal reactions, which can be highly sensitive to temperature. This is why controlling the temperature of a photochemical reaction can help the optimization phase. Temperature control can be achieved by using an immersion-well apparatus, by placing a fan in the proximity of the light source to dissipate the generated heat, or by connecting the vessel holder to a temperature-programmable device (chiller/heater) through a circulating system.
External impurities. The presence of oxygen and external impurities can also hamper the efficiency and reliability of a photochemical protocol. Oxygen can productively quench long-lived triplet-states, while short-lived excited states are less affected. Additionally, solvents and reagents are often contaminated by water and stabilizers that can interact with the photogenerated species. These effects can be bypassed using common laboratory procedures, such as Schlenk and glovebox techniques, freeze–pump–thaw degasification, inert gas sparging, and standard purification methods.
Geometry of the reaction set-up. The shape of the photochemical set-up is of primary importance. The orientation of the reaction vessel should match the illumination system to maximize the area of the reaction mixture exposed to the stream of photons. The optimal vessel positioning and distance from the light source must first be evaluated and then kept constant during the reaction optimization to ensure reproducibility. To this end, it can be beneficial to design fixed vessel holders and use reflecting surfaces (mirrors), which redirect the incident light towards the reaction solution. An additional geometrical requirement is dictated by the Beer–Lambert law (see equation and graph in Figure 2 a). According to this law, light intensity rapidly decreases towards the center of the solution. For small-scale experiments, this issue can be circumvented by appropriate stirring of the irradiated mixture. But for large-scale set-ups, the effect is amplified and drastically affects the reaction efficiency, unless compensated by an increased irradiance. Flow technologies and falling film reactors are effective solutions since they provide an almost ideal illuminated surface-to-volume ratio (Figure 2 b).24

a) The Beer–Lambert law correlates the absorbance (A) with the concentration of the chromophore (c); ϵ=molar extinction coefficient (m−1 cm−1); l=path length (cm). The light penetration decreases with distance. b) Comparison of the illumination of batch and flow reactors.
The use of flow-photoreactors brings clear benefits in terms of process scalability and productivity, and often enables shorter reaction times. Nevertheless, the use of batch systems offers lower costs and a higher operational ease for reaction monitoring/optimization. Therefore, batch apparatus could be the set-up of choice for explorative photochemical studies. As a general guideline, the scale of the photochemical process dictates the choice of reactor.25
Photochemistry is one of the branches of chemistry most influenced by technology development. It is anticipated that the ceaseless implementation of vanguard techniques, including 3D printing,26 solar concentrator reactors,27 and pulsed light irradiation,28 will provide novel opportunities to design more efficient and reliable reaction set-ups.
3 Characterization of the Ground State
The first event of any photochemical process is the absorption of light from a ground-state molecule to afford an electronically excited state. At the early stage of reaction development, mechanistic investigations should focus on the analysis of the ground-state properties of the light-absorbing molecules. The target is to identify the chromophore (to then achieve its selective and efficient irradiation) and evaluate its redox properties. This requires the chemist to study the chromatic and electrochemical features of the reaction components. Within this section, we will outline the available methods for acquiring this information, showing how this data can provide useful clues for the design and implementation of synthetic photochemical strategies.
3.1 UV-Visible (UV-vis) Absorption Spectroscopy
Empirical evaluation of the color of a substrate is the first experiment a photochemist should undertake. However, human vison detects only visible chromophores at moderate concentration and in a subjective way. An accurate analysis of the absorption spectra of the reaction components is the starting point of any photochemical methodology development. From a qualitative point of view, UV-vis spectroscopy informs about the portion of light (i.e. the range of wavelengths) absorbed by the chromophore. Quantitatively, the Beer–Lambert law (Figure 2 a) relates the absorbance (A) with the concentration (c) of the chromophore. The analysis of the absorption profile of any reaction component is fundamental to selecting the most appropriate light source and experimental set-up.
Uv-vis spectroscopy can also be useful to elucidate the mechanism of light-driven reactions, as exemplified by studies on the intermolecular enantioselective α-alkylation of aldehydes catalyzed by a chiral secondary amine of type 3 (Figure 3 a). This photochemical transformation was originally reported by MacMillan4 using a ruthenium-based polypyridyl photocatalyst29 (Ru(bpy)32+ where bpy is 2,2′-bipyridine, structure detailed in Box 2). The photocatalyst was needed to generate open-shell intermediates from electron-deficient alkyl bromides 2. An ensuing radical trap from the nucleophilic chiral enamines I, formed upon condensation of aldehydes 1 with catalyst 3, provided the α-alkylation product 4 in a stereocontrolled fashion. During later investigations on the same process, a control experiment revealed that the reaction could be efficiently conducted without the need for any external photoredox catalyst when using dinitro-benzyl bromide 2 a as the substrate.30 Since the chemistry did not proceed without visible light illumination and evidence was collected supporting an open-shell manifold, this unexpected observation posed the mechanistic conundrum of which photochemical path was responsible for the radical generation. Optical absorption spectroscopic studies of the reaction mixture led to the detection of a red-shifted band in the visible region, which was not associated with the aldehyde 1 or the dinitro-benzyl bromide 2 a (Figure 3 a). This bathochromic shift arose from the formation of a visible-light-absorbing electron donor–acceptor (EDA) complex,31 generated in the ground state upon association of the electron-rich enamine Ia and the electron-deficient benzyl bromide 2 a. Irradiation of the colored EDA complex induced an SET event, allowing access to the key benzyl radical intermediate (not shown in Figure 3 a). A stereoselective radical trap by the enamine provided the desired α-alkylation product 4 in an enantioselective fashion. In this study, UV-vis absorption spectroscopy was instrumental to the discovery that the synthetic potential of chiral enamines can be expanded into the radical domain by exploiting their photochemical activity.22

UV-vis absorption spectroscopy as a tool for mechanistic elucidation and methodology development: a) Detection of a visible-light-absorbing electron donor–acceptor (EDA) complex whose photochemical activity drives the enantioselective α-alkylation of aldehydes 1 with dinitro-benzyl bromide 2 a; the filled grey circle represents a generic bulky substituent on the chiral organic catalyst 3. b) Detection of the bathochromic shift induced by a Lewis acid 6–enone 5 coordination was crucial to developing an enantioselective catalytic [2+2] photocycloaddition reaction. c) Detection of an enhanced absorption of the chiral Lewis acid 10–substrate 8 adduct and development of a photochemical asymmetric [2+2] cycloaddition delivering the cyclobutane 11. EWG: electron-withdrawing group; TMS: trimethylsilyl.
Evaluating the absorption profile of a reaction mixture can also assist the successful development of a photochemical process. This approach has been used to address a difficult problem in enantioselective photochemistry. When it comes to enantioselective photocatalysis, the main challenge is to avoid the unselective background reaction promoted by direct irradiation of the substrates.32 A potentially useful strategy relies on the use of a chiral catalyst capable of interacting with the substrate while eliciting a bathochromic shift in its absorption. Selective excitation of this in situ generated catalytic chiral adduct can be used to trigger a stereoselective reaction, circumventing detrimental background pathways. Relying on UV-vis analysis, Bach identified a chiral Lewis acid catalyst 6 that could efficiently form the chiral adduct II upon coordination of 5,6-dihydro-4-pyridones 5 (Figure 3 b).33 This ground-state interaction induced a significant bathochromic shift (more than 50 nm) in the absorption. Selective irradiation of the complex II, by avoiding the direct excitation of 5, promoted an asymmetric intramolecular [2+2] cycloaddition.
Another strategy in asymmetric photocatalysis exploits the interaction between a substrate and a chiral catalyst, which induces a significant increase in absorbance of the substrate-catalyst adduct (Figure 3 c). This type of interaction can also be easily detected and evaluated by UV-vis analysis. This strategy served to develop a visible-light-promoted asymmetric catalytic [2+2] cycloaddition.34 The coordination of a Rh-based chiral-at-metal complex 10 to α,β-unsaturated 2-acyl imidazole derivatives 8 generated a complex III, which exhibited a 169-fold enhancement of the absorption profile compared to the non-coordinated substrate 8. This new catalytically generated species acted as an antenna to harvest the incident light and then selectively promote an enantioselective [2+2] cycloaddition with dienes 9, while suppressing the unselective background reactivity.
3.2 Electrochemical Methods
The increased internal energy of an excited-state molecule influences its ability to donate or accept electrons, making it a better oxidant and reductant than in the ground state. Although an excited-state photocatalyst can use different bimolecular mechanisms to activate substrates (e.g. energy transfer and atom transfer, see Box 1), a great portion of photocatalytic transformations are triggered by photoinduced electron transfer (PET).35 Therefore, the evaluation of the redox properties is crucial in assessing the thermodynamic feasibility of any light-driven reaction involving PET. From a practical point of view, the standard redox potential (E) of the electron donor must match the redox properties of the acceptor, thus defining the range of partners that can productively undergo the single-electron transfer (SET) step (see Practical Highlight 1).

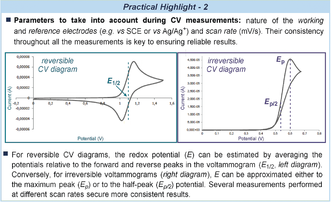
This aspect is crucial in the field of photoredox catalysis, where all the reaction components must be carefully selected to ensure the regeneration of the active photocatalyst and the quenching of the oxidized/reduced intermediates within the catalytic cycle (Figure 4 a). The literature contains standard potential values for the most common photoredox catalysts,36 several organic reagents,37 and organometallic complexes.38 However, for specific compounds or newly designed substrates, these must be obtained experimentally. Cyclic voltammetry (CV) is the most straightforward method for determining standard redox potential values (Practical Highlight 2).37, 39 Alternatively, non-electrochemical methods can be useful, including computational calculations40 and kinetic analysis via transient absorption spectroscopy.41
Electrochemical measurements can aid the selection of suitable reaction partners and guide the design of novel photoredox processes. One interesting example is Fagnoni and Albini's study of a series of benzylic silanes 12 and electron-poor olefins 13.42 The redox properties of the substrates were evaluated to identify suitable conditions for a light-driven benzylation of olefins using tetrabutylammonium decatungstenate (TBADT, structure depicted in Box 2)43 as the photoredox catalyst (Figure 4 b). The excited TBADT* triggered the oxidative cleavage of benzyl silanes 12 to generate the corresponding benzylic radicals IV. Mechanistically, because of the need to regenerate the photoactive form of TBADT (E ([W10O32]4−/[W10O32]5−)=−1.4 V vs. SCE) via an SET oxidation, it was anticipated that only easily reducible alkenes 13 could participate in the reaction. This was the exact trend observed experimentally: fumaronitrile 13 a (E (13 a/13 a.−)=−1.31 V vs. SCE) afforded the benzylation product 14 a in high yield, while acrylonitrile 13 b (E (14 a/14 a.−)=−2.17 V vs. SCE), which falls out of the redox range of TBADT, provided only trace amounts of the product 14 b.

How electrochemical methods can guide the design of a photoredox reaction: a) General mechanism of photoredox catalytic processes. It is important to match the redox potentials of the substrates and radical ion intermediates with the redox properties of the photoredox catalysts (PC). b) Selection of suitable substrates based on their redox potential: only easily reducible olefins 13, which can turn over the TBADT photocatalyst by triggering a SET oxidation of the reduced intermediate [W10O32]5−, can successfully participate in the reaction. c) Using electrochemical studies to aid substrate design: the best match between the redox properties of the photoredox catalysts and the substrates 15/16 resulted in better reactivity; Mes-Acr=acridinium salt photocatalyst (structure depicted in Box 2).
Tuning the redox properties of the substrates to enable a desired reactivity is a useful strategy when implementing a new photochemical process. The most straightforward approach relies on the modification of the electronic properties of the reagent by introducing moieties to either lower or increase its redox potential. Such modifications can be imparted either by simple backbone substitution (e.g. electron-donating groups on an arene make its SET oxidation more facile, while electron-withdrawing groups favor arene reduction) or by the use of electron-auxiliary (EA) groups.44 In addition to influencing the redox potential of the whole molecule, EA groups can undergo an irreversible fragmentation upon the SET event, ultimately facilitating the formation of reactive radical intermediates. This approach was used by Leonori to design electronically tuned imino radical precursors of type of 15 and 16, which could deliver the target iminyl radical V under photocatalytic activation (Figure 4 c).45 V could then undergo a facile 5-exo-trig cyclization onto an olefin appended within the substrate. This intramolecular step generated a carbon radical (not shown in Figure 4) that could be intercepted by different radicalophiles X–Y. The overall process led to the formation of imino-functionalized 5-membered products 17. To identify suitable iminyl radical precursors, a series of parent oximes were synthesized. O-aryl oximes 15 a–c could deliver V upon SET reduction, followed by N−O cleavage, while oximes 16 a–c, adorned with a methylenecarboxylic moiety, required a SET oxidation to generate the iminyl radical. Electrochemical investigations facilitated the identification of substrates 15 a45a (E (15 a/15 a.−)=−0.55 V) and 16 a45b (E (16 a.+/16 a)=+1.65 V) as the best radical precursors, given their match with the redox properties of eosin Y46 and an acridinium salt,47 respectively, which served as the organic photoredox catalysts.
Electrochemical investigations can also inform the selection of suitable additives that can facilitate the desired reactivity. There are cases when the direct SET between an excited photocatalyst and the substrate can be thwarted by the formation of either unstable or short-lived radical intermediates. Here, the use of electron mediators (EM) can be beneficial in facilitating exergonic redox processes that are kinetically slow, or in mitigating the occurrence of a back-electron transfer (BET) and competitive side-reactivity. Electron mediators, which are widely used in electrochemistry,48 possess stable and persistent radical ionic states and serve as electron shuttles. A recent example demonstrated how the proper choice of an electron mediator could avoid a side reaction while channeling the photochemical process towards the desired pathway. In the context of a photocatalytic styrene cyclodimerization protocol, Nicewicz used electron mediators to enable the formation of the cycloadduct 19 (Figure 5 a).49 The process was catalyzed by 2,4,6-tris(4-methoxyphenyl)pyrylium tetrafluoroborate (p-OMeTPT), which upon excitation becomes a strong oxidant (E (p-OMeTPT+*/p-OMeTPT.)=+1.74 V). p-OMeTPT* would then easily promote the SET oxidation of the alkene substrate 18 leading to the radical cation VI, which is responsible for the desired cyclodimerization. However, the excited photocatalyst is also capable of oxidizing the cycloaddition product 19, thus promoting an unproductive cycloreversion (from 19 to VI). For the reaction to selectively afford the cycloadduct 19, the electron mediator was required to have a marginally higher oxidation potential than the alkene substrate (E (18.+/18)=+1.37 V). This was the case for naphthalene (Eox=+ 1.61 V), which effectively promoted the process. A drastically lower reactivity was observed when using less oxidizing electron mediators (such as NPh3 and anthracene).

Electrochemical investigations and method development: a) guiding the choice of electron mediators (EM) and b) the design of redox stable chiral organic catalysts; TDS: thexyl-dimethylsilyl.
Electrochemical studies can also provide important mechanistic insights for optimizing a photocatalytic process. The modulation of the electronic properties of a series of chiral secondary amine catalysts 22 was key to enabling the photochemical organocatalytic asymmetric β-benzylation of cinnamaldehyde 20 with benzyl trimethylsilane 21 (Figure 5 b).50a The design of the catalyst was informed by CV analysis of the commercially available amines 22 a,b. The poor catalytic activity of the diarylprolinol silylether51 22 b was rationalized on the basis of its electron-rich nature, which imparted an oxidation potential similar to the benzyl silane substrate. This situation favored an SET oxidation of the catalyst from the strongly oxidizing photoexcited iminium ion (E (VII+*/VII.)=+2.3 V vs. Ag/Ag+ in CH3CN). This SET event resulted in an undesired catalyst degradation path. In contrast, catalyst 22 a, having a higher oxidation potential, could efficiently promote the transformation, although with low stereocontrol. Based on this observation, a novel class of aminocatalysts was designed, with modified electronic properties and enhanced oxidation potential (E (22.+/22)=+2.20 and +2.40 V vs. Ag/Ag+ in CH3CN for 22 c and 22 d, respectively), providing optimal efficiency in the photochemical asymmetric reaction. This study was instrumental for the development of other asymmetric photocatalytic methodologies exploiting the oxidizing ability of chiral iminium ions in the excited state.50b–50c
4 Characterization of the Excited State
Light absorption results in the population of electronically excited states. The photophysical processes occurring after this event are illustrated in the Jabłoński diagrams (Figure 6 a). According to the spin selection rules, light excitation generally produces a singlet-excited state (S1) that rapidly relaxes to the lowest vibrational level. S1 can then decay back to the ground state S0 through either thermal or radiative pathways. Alternatively, S1 can undergo intersystem crossing (i.s.c.) giving access to a triplet state (T1), which decays in similar ways. Excited-state entities differ fundamentally from their ground-state counterparts mainly in the energy content and the electronic distribution. Therefore, they must be treated as new chemical species with new physical properties and chemical reactivity.52 Investigating the properties of the species in the excited state is therefore fundamental to understanding and predicting how they interact with ground-state components, and which chemical processes they may trigger.

a) Jabłoński diagram; i.c.: internal conversion; i.s.c.: intersystem crossing. b) Spectroscopic determination of the energy of an excited state E00. c) Simplified Rehm–Weller equations and their use to estimate the redox potential of excited-state species (E*).
4.1 Energy of the Excited State
The energy of an excited state E00(X*/X), for a given species X, is approximated by the energy difference between the lowest vibrational level of the ground state S0 and the corresponding excited state Sn or Tn. In the case of singlet-excited state, the energy value can be obtained spectroscopically. In the absence of vibrational structures, E00 can be roughly estimated from the position of the long wavelength tail (λtail) of the absorption spectrum (Figure 6 b).52 In the case of photochemical reactions proceeding through SET, this energetic value can be exploited to estimate the redox potential of the excited-state species (E*). According to the Rehm–Weller theory,53 and when neglecting the difference in entropy between the ground and the excited state and the Coulombic interactions, a good approximation of E* can be obtained from the ground-state redox potential and the energy gap between the ground state and the excited state (E00, equations in Figure 6 c). Other approaches, including computation54 and photomodulated voltammetry,55 can be used to determine the energy of an excited state (see Practical Highlights 3 in Figure 6).
This simple method for estimating the excited-state redox potential of substrates can be useful in designing photochemical reactions. Electrochemical and spectroscopic measurements were recently applied to define the ability of dihydropyridine 24 (Hantzsch ester) to act as a photo-reductant upon excitation (Figure 7 a).56 1,4-dihydropyridine derivatives are primarily understood as hydride (H−) sources in their ground state.57 Seminal studies by Fukuzumi identified the capability of structurally related 1,4-dihydronicotinamide (NADH analogs) to reach an excited state and then reduce alkyl halides via an SET manifold.58 This precedent inspired Cho and co-workers to assess the reductive ability of dihydropyridine 24 in its excited state. The estimated high reducing power (E (24.+/24*)=−2.28 V vs. SCE) established the feasibility of triggering the photochemical reductive debromination of α-bromo ketones 25, delivering products 26. Recently, the direct photochemistry of dihydropyridine derivatives was used to design novel photochemical transformations in the absence of external photocatalysts.59

Using the excited-state energy to aid the design of new photochemical reactions: a) light excitation turns 1,4-dihydropyridines 24 into strong reductants; b) asymmetric [2+2] photocycloadditions of 2′-hydroxychalcone 27 via chiral Lewis acid-catalyzed triplet energy transfer.
The determination of the excited-state energy also provides fundamental information for the design of photoreactions proceeding through energy transfer manifolds. The triplet energy of the light-absorbing species must be higher than the triplet energy of the substrate to promote a productive sensitization. To enable this mechanism, Yoon tuned the triplet energy of 2′-hydroxychalcone 27 to facilitate a photosensitized enantioselective intermolecular [2+2] cycloaddition (Figure 7 b).60 The coordination of a chiral Lewis acid, generated from scandium triflate and a chiral ligand ((S,S)-tBu-PyBox), with substrate 27 resulted in the formation of complex VIII (ET≈30 kcal mol−1), which has a much lower triplet energy than the uncoordinated substrate (ET ≈50 kcal mol−1). This enabled a triplet energy transfer from an electronically excited racemic photosensitizer (Ru(bpy)3(PF6)2, ET=46 kcal mol−1) to VIII exclusively. This selective sensitization, ultimately catalyzed by the scandium-based chiral Lewis acid, allowed the development of an enantioselective [2+2] photocycloaddition with diene 28 to afford cyclobutanes 29 with high stereoselectivity.
4.2 Luminescence
The radiative deactivation of excited states is at the origin of the phenomenon of luminescence. This is defined as either fluorescence, when it occurs from singlet excited states, or phosphorescence, when it happens from triplet states. These two mechanisms mainly differ in their emission rate, which brings about lifetimes in the order of nanoseconds for fluorescence and micro/milliseconds for phosphorescence. Therefore, measuring the lifespan of excited states, by means of time-correlated single photon counting (TCSPC), provides a way to discriminate between singlet and triplet states.61 The analysis of the steady-state luminescence is conducted through spectrofluorometers, where the emission profile of a continuously irradiated compound is recorded. Luminescence bands generally occur at longer wavelengths than the absorption ones (Stoke shift) and the emission profile is independent from the excitation wavelength (Kasha rule, vibrational decays).62 The analysis of the emission of a fluorophore can provide useful mechanistic information. One of the most exploited and simple experiments is the luminescence quenching analysis. The decreasing of the emission intensity of an excited intermediate (quenching) can be caused by a variety of processes. One such process, the collisional quenching, occurs when an excited-state entity is deactivated by collisions with another species (quencher). Collisional quenching is governed by the Stern–Volmer equation, which relates the intensity of emission to the concentration of the quencher (see Practical Highlights 4). Accordingly, emission-quenching experiments can be used to highlight the interaction of excited-state species with other reaction components. Although this approach does not inform about the actual pathway of excited-state deactivation (e.g. it cannot discriminate between energy-transfer and electron-transfer manifolds),63 it can provide useful clues for elucidating the mechanism of a photochemical reaction.

Luminescence quenching studies were crucial to understand the mechanism of the enamine-mediated photochemical asymmetric α-alkylation of aldehydes (Figure 8 a).64 The chiral secondary amine catalyst 32 promoted the reaction of aliphatic aldehydes 30 with diethyl bromomalonate 31 under CFL irradiation, providing the corresponding α-alkylation product 33. This photochemical reaction proceeded via a radical path in the absence of any photoredox catalyst and without the formation of a visible-light-absorbing enamine-based EDA complex. To uncover the photochemical mechanism responsible for the generation of radicals, the enamine intermediate IX was isolated and submitted to Stern–Volmer quenching experiments, given its ability to emit upon excitation at 365 nm. These studies established the occurrence of a direct interaction between the excited state of IX* and the ground-state substrate 31. Following this observation, an SET from the excited enamine to the bromomalonate was proposed, which triggered the formation of a carbon-centered radical upon reductive C−Br bond cleavage. The radical was then stereoselectively intercepted by the chiral enamine, leading to the enantioenriched product 33.

Stern–Volmer quenching studies as a mechanistic probe of photochemical reactions: a) evidence that the excited enamine IX* interacts with the ground-state diethyl bromomalonate 31; b) demonstration that the emission of the iridium sensitizer 37 is quenched by the NiII species X: energy transfer from 37 produces an excited-state nickel(II) complex that couples aryl halides with carboxylic acids.
MacMillan and McCusker exploited similar collisional quenching investigations to study the mechanism of a Ni-catalyzed photosensitized coupling of carboxylic acid 34 with aryl halides 35 (Figure 8 b).65 The experiment revealed that the luminescence of the excited Ir(ppy)3 photocatalyst 37 was quenched by a NiII complex of type X. The latter is formed during the catalytic cycle upon oxidative addition of the Ni0 species into the aryl halide 35 and coordination of the metal center to the nucleophilic carboxylate. On the basis of extensive experimental investigations,63, 66 an energy transfer between the photocatalyst and the nickel intermediate was proposed as the origin of the emission quenching. This triplet sensitization of the organometallic complex X triggered the formation of an excited-state NiII species that easily underwent an otherwise thermally unfeasible reductive elimination step, eventually forging the C−O bond within product 36.
Luminescence quenching analysis can also be exploited to discover novel photochemical processes. This approach was adopted by Glorius, who recently reported a screening method to accelerate the identification of potential substrates amenable to light-driven reactions (Figure 9).67 Specifically, the emission profiles of a set of excited-state photocatalysts were recorded in the presence of several reaction candidates, randomly selected from among common organic molecules. The presence of a luminescence quenching indicated an interaction between the excited photocatalyst and the corresponding compound, representing a potential substrate amenable to a transformation catalyzed by the photoredox catalyst. Using this strategy and following critical evaluation to exclude potential issues related to the Stern–Volmer quenching experiments (e.g. inner-filter effect and static quenching, see Practical Highlight 4), the authors identified benzotriazole derivatives 38 and electron-rich phenols 39 as new classes of substrates that can productively engage in photocatalyzed protocols. This approach might be particularly interesting if integrated in the field of automation and high-throughput screening.

A screening method based on luminescence-quenching studies to aid the development of photocatalytic reactions.
5 Characterization of Reaction Intermediates
The synthetic power of photocatalysis relies on the ability to generate and harness highly reactive species, including radicals and triplet-state excited molecules, under mild conditions and in catalytic amounts. The detection and characterization of these transient intermediates is paramount for a thorough mechanistic understanding of the photochemical process. However, these studies are challenged by the fleeting nature and the short lifespan of the reactive intermediates. This section details a selection of common techniques for characterizing photochemically generated intermediates, highlighting how these analyses can help elucidate ambiguous reaction pathways.
5.1 Trapping and Quenching of Reaction Intermediates
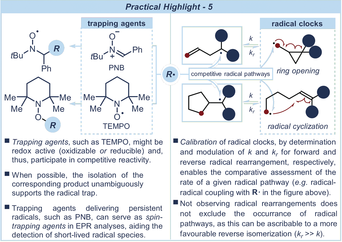
The participation of radical species in many light-driven processes creates a strong link between photochemistry and open-shell reactivity.10, 13 This is why mechanistic investigations of photocatalytic processes must often rely on strategies typical of the radical realm. The most straightforward method to ascertain the formation of radicals is to bridle them into long-lived intermediates by means of appropriate “traps” (see Practical Highlight 5). Commonly used radical trapping agents are aliphatic nitroxide derivatives, including phenyl N-tert-butylnitrone (PNB) and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO).68 Despite often being adopted for mere control experiments, radical trapping agents can also be exploited for synthetic purposes.69 An alternative strategy to evaluate the occurrence of a radical pathway relies on the use of “radical clocks”.70 These compounds are adorned with moieties that, in the presence of radicals, undergo competitive unimolecular rearrangement pathways (i.e. ring opening or radical cyclization). Radical clock compounds also provide an indirect method to determine the kinetics of a specific free-radical reaction by comparison with the known rate of the rearrangement process. For light-driven processes proceeding through energy transfer, which do not involve radical intermediates, the occurring sensitization mechanism can be probed using triplet-state quenchers, including oxygen and 2,5-dimethylhexa-2,4-diene, which act as inhibitors for the examined reaction.
5.2 Detection of Reaction Intermediates
Often, the fleeting nature of the short-lived intermediates of light-driven reactions makes their isolation/trapping unfeasible. In these cases, the participation of transients can be evaluated by their analytical detection. Due to their paramagnetic nature, free radical intermediates can be detected by electron paramagnetic resonance (EPR) spectroscopy.71 This method enables the identification of radical species through specific signal patterns, characterized by g values and hyperfine coupling constants a. Similarly to NMR analyses, EPR furnishes information on the nature and the chemical environment of a given radical. Extremely short-lived radicals can exhibit weak signals or not be observed by EPR spectroscopy. In these cases, the use of spin-trapping agents, such as PNB (see Practical Highlight 5, left side), aids the formation of more stable and detectable radicals. This strategy was recently used by practitioners of photocatalysis to detect mechanistically relevant photogenerated radical intermediates.72 NMR spectroscopy can also be used to detect the formation of radicals during photochemical reactions using Chemical Induced Dynamic Nuclear Polarization (CIDNP) spectroscopy.73 This technique can detect the “signatures” left by short-lived radical pairs in the nuclei of the downstream diamagnetic species, which are detectable by simple 1H NMR analysis in the form of enhanced absorption and emission peaks. This method is independent of the radical lifetimes, and short-lived open shell species are detectable in the minute timescale. Generally, a photo-CIDNP experiment requires in situ illumination within the NMR spectrometer. Easily assembled LED-based devices, which can be used to provide high photon flux directly in the NMR tubes, have been reported74 and used to elucidate photochemical reaction mechanisms.75
The detection of highly elusive intermediates, including non-radical ones, requires more sensitive time-resolved spectroscopic techniques,76 such as laser flash photolysis (LFP).77 These methods enable the observation of species with lifespans in the order of pico/femto-seconds, including molecules in their triplet/singlet excited states and transient ionic radicals. In LFP, an analyte mixture is exposed to intermitting irradiation by an ultrafast pulsing laser. The difference in absorbance or emission before and after light collision (the optical deviation, ΔOD) is recorded. Figuratively speaking, LFP analyses provide a snapshot and a biography of photogenerated intermediates. Specifically, LFP curves against wavelength (λ) give information on the presence/nature of an intermediate (Figure 10 a, left diagram). In contrast, recording the LFP curve over time, at a specific wavelength, describes the evolution/disappearance of a specific intermediate (Figure 10 a, right diagram).

a) Laser flash photolysis (LFP) can provide information on the formation (ΔOD vs. λ) or decay (ΔOD vs. t) of a transient intermediate. b) Application of LFP to elucidate the mechanism of the anti-Markovnikov alkene hydrofunctionalization catalyzed by the acridinium salt (Mes-Acr+).
Nicewicz78 used the first approach (ΔOD vs. λ) to detect the transient radical cation XI. This intermediate was supposed to play a crucial role in the photocatalytic oxidative hydrofunctionalization of alkenes 40 catalyzed by the acridinium salt (Mes-Acr+),47 affording the anti-Markovnikov adducts 41 (Figure 10 b). Through LFP analysis of a solution of the photoredox catalyst and anethole 40 a, the authors could identify the absorption profile of the radical cation XIa (peak at ≈600 nm, purple line). This was obtained by subtracting the UV-vis spectrum of the reduced state of the photocatalyst (Acr-Mes., dashed red line), independently registered after electrolysis of Acr-Mes+, from the LFP curve (light blue line). The data obtained for XIa were in accordance with those available in the literature for structurally similar styrenyl cation radicals, thus supporting the formation of the radical cation under the reaction conditions. This finding validated the originally postulated mechanism proceeding through SET oxidation of 40 by the acridinium catalyst.79
6 Kinetic Studies
A large number of photocatalytic transformations exploit redox processes. As discussed in Section 3.2, redox potentials are useful in assessing the thermodynamic feasibility of a reaction. But the viability of a process also depends on kinetic parameters. This is particularly true in photochemical and radical processes, where the fleeting nature of excited-state transients and open-shell reactive intermediates limits their ability to productively engage in bimolecular processes.
6.1 Excited-State Kinetics
Stern–Volmer analysis is a widely applied method for studying the excited-state kinetics of an emissive molecule (see Practical Highlight 4 and Section 4.2). Steady-state luminescence quenching studies can provide useful information on the kinetics of bimolecular events, such as the rate constant of dynamic quenching processes. Non-emissive excited-state transients create a more difficult situation for kinetic analysis.14a, 77a In this instance, LFP analysis can inform about the rate of formation or decay of fleeting photogenerated intermediates (Figure 10 a, ΔOD vs. t). A detailed understanding of the excited-state kinetics that govern the dynamics of the photocatalytic process can be instrumental to design and optimize a synthetic methodology. This was recently demonstrated by Scaiano, who used kinetic investigations via transient absorption spectroscopy to develop a metal-free variant for a photocatalytic oxidative hydroxylation of aryl boronic acids 42 with molecular oxygen (Figure 11 a).80 This process was previously reported using [Ru(bpy)3]Cl2 as the photoredox catalyst, but it suffered from long reaction times.81 The excited-state decay of [Ru(bpy)3]Cl2 and methylene blue (MB), an organic triplet photosensitizer, were monitored by LFP in the presence of increasing concentrations of N,N-diisopropylethylamine as sacrificial quencher. The bimolecular quenching constants (kobs) were plotted as a function of the quencher concentration, allowing the identification of MB as the most efficient photocatalyst (MB was quenched by iPr2NEt ca. 40 times faster than [Ru]). Experimentally, this translated to a faster reactivity observed for the oxidation reaction catalyzed by MB, which delivered phenol 43 in 94 % yield after 7 hours. In contrast, when the Ru-based photocatalyst was used, product 43 was obtained in 54 % yield after the same time.

a) Laser flash photolysis (LFP) can provide information about the excited-state kinetics (ΔOD vs. t). Here this approach guided the identification of a more efficient protocol for the oxidative hydroxylation of aryl boronic acids. b) Initial-rate kinetic analysis for the rate-order assessment and the identification of the turnover-limiting step of a photocatalytic reaction.
6.2 Kinetics of the Overall Process
Approaches based on time-resolved spectroscopic methods allow the measurement of the absolute rate constants of individual steps of a photochemical process. This approach is commonly used in radical chemistry, where clock methodology (see Practical Highlight 5) serves to evaluate individual rate constants of all the steps in a process. Recent studies have suggested that the method of initial-rate measurements, mutated by the polar ground-state reactivity domain, can provide useful kinetic information about the overall photochemical and radical transformations. This approach was used to identify the turnover-limiting step of a TBADT-photocatalyzed enantioselective radical conjugated addition to cyclic enones 44 (Figure 11 b).82 Upon light excitation at 365 nm, the photocatalyst generated radicals from precursors 45 via a hydrogen-transfer mechanism (HAT). The use of the chiral amine catalyst of type 46, adorned with a redox-active carbazole unit, was crucial to the stereocontrolled formation of products 47.83 The organic catalyst's effect on the reaction efficiency was evaluated with a set of newly synthesized carbazole-containing amines 46 a–e, with a wide range of electronic properties but a comparable steric-shielding ability. Preliminary kinetic experiments, conducted by NMR spectroscopy, aimed to evaluate how the light intensity influenced the rate of reaction. A light-saturation regime was reached when applying an irradiance of ≈40 mW cm−2, thus providing reliable conditions for maximizing the amount of excited TBADT in solution. The importance of creating a photon-limited regime in initial-rate kinetic analysis was also highlighted by Nicewicz, who used a similar kinetic treatment to study photocatalytic hydrodecarboxylation reactions.84 Under this illumination condition, kinetic experiments indicated a linear correlation between the observed initial rates and the redox potentials (Ered) of the carbazole catalysts 46 a–e: the larger the reduction potential of the carbazole tethered to the aminocatalyst, the faster the reaction proceeded. A reaction-profile analysis was then undertaken to assess the rate-order dependence, which established a first order in the catalyst, a zeroth order in radical precursor 45, and a saturation kinetic profile for both the enone 44 and TBADT photocatalyst. Collectively, these findings were consistent with the notion that the overall turnover-limiting step is the SET oxidation of TBADT-H (the reduced form of the photocatalyst) by the long-lived carbazoliumyl radical cation in XII to return both the neutral carbazole catalyst in XIII and the ground-state photoredox catalyst TBADT. In consonance with this mechanistic framework, the carbazoliumyl radical cation XII was detected by visible absorption spectrophotometry in the reaction mixture, thus indicating that this species accumulates before the reaction's slowest step. This kinetic analysis provided crucial information to design a more effective carbazole-based catalyst of type 46 for the development of a new enantioselective photochemical reaction.85
7 Quantum Yield Determination
Often, photochemical reactions generate radicals that are responsible for product formation. Some processes proceed via a radical-radical coupling mechanism, where open-shell intermediates combine to afford a closed-shell species. This requires at least one of the radicals to be persistent (the persistent radical effect),86 or that radicals are generated at a similar rate and in close proximity. In many cases, radical reactions proceed through self-propagating radical-chain pathways.87 In such processes, the product is formed through propagation steps that convert the radical, (not necessarily) photochemically generated from the substrate precursor, into the final product while regenerating the chain-propagating radical. In chain processes, the photochemical step would therefore serve as an initiation event that generates the original radical. To discriminate between a radical coupling and a self-propagating chain mechanism, chemists have often relied on intermittent irradiation with on/off times (light off/light on experiment). Despite its practical ease, this approach is not useful since chain reactions are generally terminated milliseconds after illumination is turned off. This experiment only offers qualitative evidence that light is necessary to trigger the reaction.

A proper method of probing a chain-propagating mechanism is to determine the quantum yield (Φ) of the light-induced reaction (refer to equation in Practical Highlight 6). For a process in which one photon produces only one molecule of the product, the quantum yield can be maximum 1. For a chain process, where one photon forms n molecules of product, the quantum yield is expected to be >1. But a value of Φ<1 does not exclude a possible radical chain mechanism, since it can be a consequence of a highly inefficient initiation step. Yoon used quantum yield determination to evaluate the mechanism of a set of previously reported radical processes, all catalyzed by [Ru(bpy)3]2+ (Figure 12 a).89 In the first case, the photocatalytic [4+2] cyclization between alkene 48 and diene 49, delivering cycloadduct 50, proceeds through the radical cation XIV. The radical anion XV is a key intermediate in the photocatalyzed [2+2] intramolecular cyclization of enone 51 to afford the cyclic product 52. Finally, the photoinduced formation of the neutral radical XVI from bromomalonate 54 enables the asymmetric organocatalytic α-alkylation of aldehydes 53. In all cases, a quantum yield higher than 1 was obtained, supporting the involvement of a self-propagating radical chain mechanism as the main reaction pathway.

a) Characterization of chain processes in ruthenium-catalyzed photochemical processes. b) Intermittent illumination to study a ruthenium-catalyzed photochemical process.
Recently, Scaiano reported the development, characterization, and calibration of a novel actinometer based on the widely used photocatalyst [Ru(bpy)3]Cl2.90 This system, which simplifies quantum yield determination, has the potential to be widely adopted by the synthetic photochemistry community. The same study90 demonstrated the possibility of using light-pulsed technology to characterize light-induced radical chain reactions. This method implements the rotating sector (RS) approach used in the past91 to translate the light-on/light-off experiment on a millisecond time-scale, overall allowing detection of very short-lived chains.45 The photocatalytic oxidation of benzhydrol 56 in the presence of 4-cyano-N-methoxypyridinium 57 was studied (Figure 12 b). Running the reaction under pulsed LED irradiation and changing the frequency of the intermittent illumination provided the desired product 58 in different yields, thus proving the involvement of a chain propagation mechanism. Data treatment provided an estimation of the radical chain lifetime (τs=19 ms).
8 Conclusion
“Man, being the servant and interpreter of Nature, can do and understand so much and so much only as he has observed in fact or in thought of the course of nature: beyond this he neither knows anything nor can do anything” Francis Bacon (1561–1626)
Understanding and explaining an observed phenomenon is the goal of the scientific method. This applies also to chemistry. Since its foundation, the discovery of novel chemical transformations has prompted theories to explain the underlying mechanisms. The field of synthetic photocatalysis has recently experienced an incredible expansion because of its intrinsic synthetic potential. This has led to the invention of many unprecedented photochemical methodologies in a relatively short period of time. However, the feeling remains that the pace of this growth has somehow increased the gap between experimental results and mechanistic understanding.
This Review seeks to highlight how mechanistic investigations can be instrumental in guiding the invention and development of synthetically useful photocatalytic transformations. We provide a simplified roadmap (Figure 13) that can be used by synthetic photochemists to temporally identify the proper mechanistic experiments to perform at different stages of methodology development. We have collected successful examples in which mechanistic studies have been essential to successfully developing light-triggered transformations. Mechanistic investigations can play a decisive role throughout a methodological project, from the design of a novel reaction to the mechanistically informed development of the method. Therefore, chemists should avoid relegating these investigations to the project's final phase. In addition to the aesthetic aspect that an elegant and adequately supported mechanistic proposal adds to a synthetic photochemical study, the characterization of excited- and ground-state intermediates along with the identification of transient species provides fundamental information for fully exploiting newly identified reactivity concepts. We expect that the combination of experimental and mechanistic studies will play an increasingly dominant role in sustaining further innovation in the field of photocatalysis.

a) A roadmap for assisting reaction development in photocatalysis using mechanistic studies.
Acknowledgements
We thank MICIU (CTQ2016-75520-P) and the European Research Council (ERC 681840—CATA-LUX) for financial support. LB thanks MICIU for a predoctoral fellowship (CTQ2013-45938-P). G.E.M.C. thanks the Marie Skłodowska-Curie Actions for a postdoctoral fellowship (H2020-MSCA-IF-2017 795793). P.M. thanks the past and present members of the group for invaluable discussions about mechanisms and photocatalysis.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Luca Buzzetti was born in Morbegno, Italy, in 1990. He studied chemistry at the University of Pavia, where he obtained his MSc degree in 2014 (Prof. Maurizio Fagnoni as the supervisor). He is currently a PhD candidate in the group of Prof. Paolo Melchiorre at the Institute of Chemical Research of Catalonia (ICIQ), Tarragona (Spain). His research interests focus on the development of photochemical reactions for the formation of C−C bonds.

Biographical Information
Giacomo E. M. Crisenza was born in Trezzo sull'Adda, Italy, in 1988. He completed his undergraduate studies at the “Università degli Studi” in Milan. He conducted his thesis projects in collaboration with Prof. Dalla Croce and Prof. La Rosa, obtaining his Master's degree in Organic Chemistry in 2012. In the same year, he joined the Chemical Synthesis CDT programme at the University of Bristol, where he undertook his doctoral studies under the supervision of Prof. John F. Bower. In 2017, he joined the group of Prof. Paolo Melchiorre at ICIQ, Tarragona, where he is a Marie Skłodowska-Curie postdoctoral fellow.

Biographical Information
Paolo Melchiorre was born in 1973 in Italy. He earned his MSc (1999) and PhD (2003) in Chemistry from Bologna University under the supervision of A. Umani-Ronchi and P. G. Cozzi. After a research period with K. A. Jørgensen, Århus University (Denmark), he joined G. Bartoli's research group at Bologna University, where he became Assistant Professor in 2007. In 2009, he moved to the ICIQ, Tarragona (Spain) as an ICREA Research Professor. In 2018, he was appointed as a Senior Tenured Scientist at the Italian Institute of Technology IIT, Genoa (Italy).


