

From Chemical Topology to Molecular Machines (Nobel Lecture)†
Copyright© The Nobel Foundation 2016. We thank the Nobel Foundation, Stockholm, for permission to print this lecture.
Graphical Abstract
Magic rings: The field of molecular machines has its origins in the synthesis of catenanes and rotaxanes. J.-P. Sauvage describes in his Nobel Lecture the beginnings of this research and the developments that led to the first molecular muscles and machines whose movement can be directed “from the outside” in a controlled manner.

Abstract
To a large extent, the field of “molecular machines” started after several groups were able to prepare, reasonably easily, interlocking ring compounds (named catenanes for compounds consisting of interlocking rings and rotaxanes for rings threaded by molecular filaments or axes). Important families of molecular machines not belonging to the interlocking world were also designed, prepared, and studied but, for most of them, their elaboration was more recent than that of catenanes or rotaxanes. Since the creation of interlocking ring molecules is so important in relation to the molecular machinery area, we will start with this aspect of our work. The second part will naturally be devoted to the dynamic properties of such systems and to the compounds for which motions can be directed in a controlled manner from the outside, that is, molecular machines. We will restrict our discussion to a very limited number of examples which we consider as particularly representative of the field.
Chemical Topology
Generally speaking, chemical topology refers to molecules whose graph (i.e. their representation based on atoms and bonds) is nonplanar.1, 2 A nonplanar graph can not be represented in a plane or on a sheet of paper without crossing points. In topology, the object can be distorted as much as one likes, but its topological properties are not modified as long as no cleavage occurs.3 In other words, a circle and an ellipse are topologically identical. The most representative examples of topologically nonplanar, and thus nontrivial, molecules are interlocking ring molecules or knotted cycles such as the trefoil knot. Interestingly, topology and chemistry were not at all connected until 1961, when two chemists working at Bell Telephone Laboratories, Frisch and Wasserman, published an important discussion simply entitled “Chemical Topology”.1 For the first time, chemists were exposed to topology thanks to this seminal paper. It contains a very clear account of most of the ideas that constitute the background of chemical topology. For example, the idea of topological isomers (also named “stereoisomers”) is introduced by comparing a [2]catenane (drawing III, two interlocked rings) to the set of the two separate cyclic molecules (object IV), as shown in Figure 1. Another example is provided by considering a single closed curve, which can be a normal cycle such as I (topologically trivial or planar) or the knotted cycle, the simplest example being the trefoil knot II. Hypothetical compounds I and II of Figure 1 are topological stereoisomers: although they may consist of exactly the same atoms in the same sequence, and the same chemical bonds between them, they cannot be interconverted by any type of continuous deformation in three-dimensional space.

The historical publication introducing the notion of chemical topology in the chemical literature. Trefoil knot II and catenane III are topologically nonplanar in the sense that their graphs, once projected on a plane, will always require crossings for being represented.
Schill's book “Catenanes, Rotaxanes and Knots”, written in 1971, is indispensable for the topologist.4 In addition to interesting theoretical considerations regarding interlocked rings and knots, it contains much information on the experimental approaches used by Lüttringhaus, Schill, and their co-workers to prepare such topologically novel systems. One of the first [2]catenanes to be reported in the literature was prepared by Schill and Lüttringhaus more than 50 years ago (Figure 2).5 This remarkable piece of work was greeted by the scientific community due to the elegance of the procedure and also due to the impressive synthetic work it represented at the time. Unfortunately, the complete synthesis required a large number of steps and, as a consequence, the overall yield was limited. Despite the novelty of the work, there were very few follow-up contributions from other organic chemists, mostly because of the extreme difficulty that a gram-scale synthesis of such compounds would represent following a purely organic chemistry approach similar to that of Schill and Lüttringhaus.

A [2]catenane and a [2]rotaxane. The number in square brackets (2 in this case) indicates the number of components incorporated in the interlocking structure. A [2]catenane is considered as the prototype of molecules having a nonplanar graph. In mathematics, the object consisting of two interlocking rings is called a “Hopf link”. Heinz Hopf was a German topologist.
Before our group started in the field of catenanes, we were interested in markedly different topics. We contributed to various research areas, such as homogeneous catalysis of the water-gas shift reaction using iridium complexes,6 electrochemical reduction of carbon dioxide,7 and inorganic photochemistry8 in relation to solar-energy conversion into chemical energy. Similar to a relatively large number of other research teams, we were fascinated by the “grand project” known by the community as “water splitting”, that is, the photochemical splitting of the water molecule to oxygen and hydrogen, supposedly the ideal fuel for the future. Since the Nobel Foundation recommended that the laureates tell as honestly as possible “the story behind the discovery”, I would like to explain how we moved from inorganic photosensitizers to catenanes and related species. At the end of the 1970s, there was considerable interest in ruthenium complexes of the Ru(bipy)32+ family (bipy=2,2′-bipyridine).9 Several prestigious photochemistry groups had shown that this compound displays promising properties in terms of ability to transfer electrons, positive holes, or electronic energy. Even more spectacular, several approaches to the “water splitting” reactions had already been reported.10 A particularly interesting project was to replace the second-row ruthenium metal, notoriously known to be rare and expensive, by common first-row transition metals. Along this line of research, very few candidates appear to be reasonable. The only one was copper(I), since this metal was known to form highly colored complexes with aromatic diimine ligands of the bipy family. The absorption band responsible for the color in the visible spectrum was shown long ago to be a metal-to-ligand charge-transfer band. An American photochemist, David R. McMillin (Purdue University), had already published seminal work on the photochemical properties of copper(I) complexes in 1978.11 At the beginning of the 1980s, our group prepared 2,9-diphenyl-1,10-phenanthroline (dpp) for various reasons, mostly in relation to sterically hindering ligands able to favor coordination unsaturation once complexed to rhodium or iridium centers.6 David spent some time in Strasbourg when he was on sabbatical leave. He visited our group and, after several discussions, we embarked on a common project dealing with Cu(dpp)2+ and related complexes containing two intertwined ligands disposed more or less orthogonally to one another once coordinated to the central CuI. At that time, Cu(dpp)2+ was unknown, since dpp itself seemed to be a new compound. The beginning of this collaboration coincided with another major event, which was the arrival of Christiane Dietrich-Buchecker to our team. Christiane had a superb organic chemistry background and we knew that she had all the necessary expertize for making novel ligands, either by herself or associated with the PhD students she would co-supervise with me. Coming back to Cu(dpp)2+, by looking carefully at its three-dimensional representation8 as shown on Figure 3, it was relatively easy to figure out that this compound, with its two intertwined ligands, was the ideal precursor to a [2]catenane.

The simple coordination chemistry reaction leading to Cu(dpp)2+ and its three-dimensional representation.
There is perhaps an important factor in the fact that we recognized the link between Cu(dpp)2+ and catenanes: the molecules were drawn manually using Indian ink, since this period corresponded to the “pre-ChemDraw era”. We thus had time to completely “digest” the molecular structures of the compounds we were making and first had to draw. From the drawing of Figure 3, it is clear that by connecting the two para-positions of the phenyl nuclei attached to the same 1,10-phenanthroline group, one should obtain a ring. Furthermore, by doing this operation simultaneously on both dpp subunits within the complex, formation of a [2]catenane appeared to be almost certain.
With Christiane, we thus embarked in the new field of catenanes. After a few months, we obtained the first positive results and about one year after the beginning of the project, the first communication on our new strategy for making interlocking rings was published12 (Figure 4).

The title and the abstract of the first publication on catenanes from our group, with the picture of the late Dr. C. O. Dietrich-Buchecker (1942–2008).
We described this new strategy as a three-dimensional template synthesis around a transition-metal center by reference to classical template effects so efficiently used for making macrocycles by several groups, such as Busch and his team in particular.13 The work seemed to be the first practical synthesis of a [2]catenane in the sense that, for a good chemist, 500 milligrams could be obtained within a few weeks. After the preparative work performed by Christiane, it remained to characterize the compound and convince ourselves that the molecule obtained was indeed a [2]catenane. 1H NMR studies demonstrated that the two dpp coordinating fragments were intertwined, thus indicating that the rings were interlocking with one another. The general strategy is depicted in Figure 5.

Synthesis of interlocking ring systems. The strategy is based on a 3D-template effect induced by a transition-metal center (copper(I) in the present case). The arcs of a circle and the rings contain coordinating fragments f-f able to interact with a transition-metal center (black disk). The f and g letters represent chemical functions able to react with one another and form an f-g chemical bond.
Strategy A is both very general and simple. Both coordinating fragments have to bind to the metal center so as to be mutually perpendicular. By an appropriate choice of metal, chelates, linkers (g-g), and functional groups (f and g), the system consisting of two intertwined organic fragments interacting with the central metal will react in the expected fashion with formation of two interlocked rings. The only apparent weakness of strategy A is that a total of eight reacting points have to find one another in the double-cyclization reaction. The second approach, strategy B, reduces this problem since it involves only four reacting groups to be interconnected in a single cyclization (twice g-f) leading to the catenane. The only requirement is that the starting macrocycle has to be presynthesized before the template reaction is performed. This ring contains a coordinating fragment (f-f) and a noncoordinating linker (g-g), and it is also likely to be formed as an intermediate in strategy A. It is noteworthy that, provided the threaded bischelate complex (bottom left) is stable enough, it must form quantitatively from a 1:1:1 mixture of the macrocycle, the metal ion, and the open-chain fragment. A statistical mixture of complexes, as would arise if two different open-chain ligands were reacted with a metal ion, cannot occur because it is impossible for two of the macrocyclic ligands to attach to the same metal ion.
Since it was less risky, we started with strategy B, which led to an interlocked system in good yield (1983).12 Later on, we used strategy A since it is slightly shorter than strategy B in terms of number of steps from commercially available compounds (1984).14 The actual reactions carried out are shown in Figures 6 and 7. It is noteworthy that strategy B is more general than strategy A, since it opens the way to asymmetric [2]catenanes, that is, compounds containing two different rings. In addition, the threaded intermediate is close to a [2]rotaxane: it suffices to end-functionalize the threaded fragment of the compound using bulky groups to generate such a [2]rotaxane. We have used this synthesis route extensively for making porphyrin-containing catenanes and rotaxanes as well as variously substituted compounds of the same family.

“Entwining” two ligands around a copper(I) center. The starting compound represented on the left is formed from commercially available 1,10-phenanthroline in two steps. It can be made at the 10-gram scale within one or two weeks. In the presence of copper(I), the entwined compound represented on the right is formed quantitatively.

The double cyclization reaction leading to the [2]catenane is relatively low-yielding. Nevertheless, it was possible to prepare batches of 0.5 g within a few weeks. By slightly modifying the structure of the starting molecules and by applying the ring-closing methodology brilliantly proposed by Grubbs, we substantially improved the yield (up to 92 %).15
The copper-complexed catenane of Figure 7 could be quantitatively demetalated using a cyanide salt. The back reaction leading to the starting copper(I) complex from the metal-free compound was perfectly reversible, as shown in Figure 8. X-ray quality crystals of both forms could be grown and their crystallographic structures were obtained by Pascard and co-workers.16 We coined the terms “catenates” and “catenands” for complexes whose ligands are interlocking rings and for the corresponding metal free-compounds, respectively.

The demetalation/remetalation reaction and the X-ray structures of the copper(I) catenate and the catenand.
The X-ray structures are represented below the corresponding chemical drawings of the copper-complexed and metal-free [2]catenanes. As far as the copper complex is concerned, the similarity between the drawing and the X-ray structure is striking. By contrast, the shape of the solid-state free catenane is significantly different from the drawing. In any case, demetalation of the complex or complexation of the free form results in a complete rearrangement of the organic backbone. This metamorphosis can clearly be connected to the molecular machinery field, although it is still relatively far from movements taking place in real molecular machines.
The Strasbourg approach to catenanes represented a significant progress in the catenane and rotaxane field. All of sudden, the transition-metal-templated approach developed by our team opened the way to novel topologies and to rotaxanes. In fact, the potential of transition metals in relation to catenanes, rotaxanes, and even knotted rings appeared to be limitless. Although the contribution of Lüttringhaus and Schill was conceptually important, the template approach completely modified the way molecular chemists looked at catenanes. Within a few years, they changed status from very exotic and almost impossible-to-make species, at least at a macroscopic scale, to normal and reasonably accessible compounds. A few pioneering contributions based on organic templates were successfully explored by other groups. Stoddart and his group reported in 1989 a particularly efficient and attractive synthesis of a [2]catenane.17 They took advantage of acceptor–donor interactions between an electron-deficient 4,4′-bipyridinium derivative and an aromatic electron donor, combined with favorable hydrogen bonds, to predispose the various groups in a very favorable situation before a cyclization reaction leading to the desired [2]catenane. This seminal piece of work was followed by two reports based on hydrogen bonding only, published in 1994 by Hunter and his group18 and Voegtle and co-workers.19 Both teams reported almost simultaneously very similar data, although they were working far away from one another. These two pioneering hydrogen-bonding-based approaches were followed by remarkable work performed by Fujita and co-workers.20 It was based on kinetically labile Pd−N bonds and hydrophobic interactions. Fujita introduced the concept of “magic rings”, since the [2]catenane could be reversibly dissociated to the set of two non-interlocking rings by just changing the nature of the medium in which the catenane was dissolved or the concentration of its constitutive components.
As far as rotaxanes are concerned, cyclodextrins turned out to be particularly interesting. These natural rings were used as the cyclic components of a large variety of rotaxanes. In the presence of appropriate string-like organic fragments, threading through the cyclodextrin was shown to take place thanks to hydrophobic forces. The first convincing example of a cyclodextrin-based [2]rotaxane was reported by Ogino in 1981.21 This pioneering publication was followed by remarkable work from various groups and, in particular, from Harada and co-workers.22
Figure 9 gathers a few remarkable topologies which could be obtained at the molecular level by our group using a generalized transition-metal-based strategy, that is, by assembling two or more transition-metal centers and molecular threads containing two or more coordinating fragments: a [3]catenane,23 the trefoil knot,24 and a doubly interlocking [2]catenane also known as the Solomon rings.25 Other groups have also been interested in chemical topology. Impressive topologies at the molecular level have been reported by the research teams of Stoddart, Leigh, and Siegel, just to cite a few.26-28

From a simple [2]catenane to more complex topologies. The examples represented are those made in Strasbourg.
Molecular Machines
The field of molecular machines, mostly initiated by Stoddart, Balzani, and their co-workers,29, 30 was to a large extent an extension of that of interlocking ring molecules, at least at the beginning. The structures of catenanes and rotaxanes seem to be ideally suited to the making of molecular switches and machines, that is, compounds which can undergo large-amplitude motions in a controlled and reversible way (Figure 10).

Catenanes and rotaxanes in motion: towards molecular machines. Catenanes and rotaxanes are very well adapted to large-amplitude motions: a ring can glide along the axis on which it is threaded (linear motor); it can also “pirouette” around the axis or within another ring.
Electrochemically Driven Swinging Motion of a Copper-Complexed Catenane
The very first system made by our group for which a large-amplitude motion can deliberately be triggered by an external signal was reported in 1994.31 A ring could be forced to rotate inside the other ring by playing with the copper(I)/copper(II) redox couple, which led us to name the compound a “swinging catenane”. Since that time, molecular machinery has blown up in a very impressive way thanks to the great contributions of various groups, either working with catenanes or rotaxanes, or using non-interlocking compounds. Stoddart, Kaifer, and co-workers reported their first molecular “shuttle” in the same year.30 The compound made and studied in Strasbourg is a copper-complexed [2]catenane, displaying two very distinct coordination modes corresponding to two extremely different geometries. Having taken advantage of the roughly tetrahedral arrangement of the copper(I) center in bis(diimide) complexes to construct catenanes and molecular knots, the electrochemical properties of related copper complexes have later on been used in our group to design and prepare a large variety of catenanes and rotaxanes and for investigating their controlled dynamic properties, playing on the two classical oxidation states of copper, +1 and +2. As far as the first “swinging catenane” is concerned, the interconversion between both forms of the complex is electrochemically triggered and corresponds to the sliding motion of one ring within the other. It leads to a profound rearrangement of the compound and can, thus, be regarded as a complete metamorphosis of the molecule. The principle of the process is explained in Figure 11.

Principle of electrochemically induced molecular motions in a copper-complexed [2]catenane. The stable four-coordinate monovalent complex (top left, the black circle represents CuI) is oxidized to an intermediate tetrahedral divalent species (top right, the white circle represents CuII). This compound undergoes a complete reorganization process to afford the stable five-coordinate CuII complex (bottom right). Upon reduction, the five-coordinate monovalent state is formed as a transient (bottom left). Finally, the latter undergoes the conformational change that regenerates the starting complex. The bidentate coordinating fragment is phen (1,10-phenanthroline), part of the dpp ligand, and the tridentate ligand is terpy (2,2′:6′,2′′-terpyridine).
The key feature to the transformation is the difference in preferred coordination number (CN) of the two different redox states of the metal: CN=4 for copper(I) and CN=5 (or 6) for copper(II). The organic backbone of the asymmetric catenate consists of a dpp bidentate chelate included in one cycle and, interlocked to it, a ring containing two different subunits: a dpp moiety and a terpy ligand. Depending upon the mutual arrangement of both interlocking rings, the central metal copper can be tetrahedrally complexed (two dpp units) or five-coordinate (dpp+terpy). Interconversion between these two complexing modes results from a complete pirouetting of the two-site ring in a given direction or the other. It can easily be induced electrochemically, by means of a chemical reductant or oxidant, or even photochemically.32 From the stable tetrahedral monovalent complex, oxidation leads to a four-coordinate CuII state, which rearranges to the more stable five-coordinate compound. The process can be reversed by reducing the divalent state to the five-coordinate CuI complex, obtained as a transient species before a changeover process takes place to afford back the starting tetrahedral monovalent state. The real molecules are represented in Figure 12 as well as the square scheme interconverting the four- and the five-coordinate species.

Square scheme corresponding to the process whose principle is depicted in Figure 11, with the chemical structures of the compounds. The dpp-containing cycle is a 30-membered ring, whereas the dpp+terpy-incorporating one is a 31-membered ring. It could be shown that the five-coordinate CuII complex has a square-pyramidal geometry and not a trigonal-bipyramidal one.33
Interestingly, the transformations of Figure 11 and Figure 12 are accompanied by a change in the electrochemical properties of the complex as well as spectroscopic changes. As expected, the tetrahedral copper complex has a relatively high redox potential: E0=+0.63 V vs. SCE in CH3CN, whereas the five-coordinate species has a slightly negative potential, pointing to greater stabilization of the divalent copper than in the four-coordinate species: E0=−0.07 V.
The obvious weak point of the swinging catenane of Figures 11 and 12 is its kinetic inertness. It should be noted that this first system was dramatically improved by modifying structural parameters of the compounds and, in particular by replacing the [2]catenane organic backbone by a [2]rotaxane, either acting as a pirouetting species or as a two- or three-station molecular shuttle. The rearrangement process was also markedly sped up by modifying the structure of the bidentate chelate incorporated in the rings. By replacing the highly sterically hindering dpp fragment by an endocyclic but nonsterically hindering chelating group of the 8,8′-diaryl-3,3′-biisoquinoline family, fast moving rotaxanes were obtained.34, 35 Finally, it should be noted that the motion taking place in the swinging [2]catenane is by no means a rotation motion. The ring that incorporates both a bidentate and a tridentate chelating group undergoes a “pirouetting” motion corresponding to a 180° rotation in a given direction or in the other. Real rotary machines were reported much later. Feringa's contribution to the field of rotary motions was indeed crucial.36
Towards Synthetic Molecular Muscles: Contraction and Stretching of a Linear Rotaxane Dimer
The topic of this paragraph is dealing with a special type of molecular machine related to contraction and extension of switchable species reminiscent of muscles.37, 38 Rotaxane dimers as represented in Figure 13, can be regarded as synthetic analogues of natural muscles. Since real muscles mostly consist of filaments able to glide along one another, a molecular assembly in which two string-like molecular fragments can glide along one another was designed. This is the process taking place in the sarcomere, in which the thick filament (containing myosin) moves along the thin filament (actin polymer) in one direction or the other so as to induce contraction or elongation. The actin–myosin complex as well as other motor proteins are fascinating biological systems which represent an inexhaustible source of inspiration for synthetic chemists.

A synthetic “molecular muscle” [2]rotaxane dimer. The molecular structure of the rotaxane dimer or pseudo-[2]rotaxane dimer (i.e. without bulky stoppers at the ends of the threaded filaments) is adapted to the contraction/extension motion mimicking the dynamic properties of a muscle.
After careful design mostly based on space-filling models, we started the synthesis work. It turned out to be particularly delicate and time-consuming. In the present discussion, we will skip the preparation of the ring-and-lateral thread conjugate, although the synthesis of this compound was far from trivial.
A 31-membered ring incorporating a dap fragment (dap=2,9-diaryl-1,10-phenanthroline), prepared in several steps from commercially available compounds, was covalently linked to a linear fragment incorporating a 2,9-dimethyl-3,8-bis-(p-hydroxyphenyl)-1,10-phenanthroline so as to afford the bischelating target consisting of a coordinating ring attached to a lateral arm containing another bidentate chelating group. The reaction of the ring-and-string conjugate with a stoichiometric amount of [Cu(CH3CN)4]PF6 in CH3CN/CH2Cl2 at room temperature (Figure 14) led to the doubly threaded species whose structure is represented in Figure 15. The success of this latter reaction was certainly far from being certain, considering the number of CuI complexes which could be formed by coordinating the two different chelating fragments (endocyclic and lateral coordination sites) of the ligand to metal centers. Nevertheless, we were happy to observe that formation of the doubly threaded complex was quantitative. In fact, the complexity of the reaction leading to the doubly threaded species is reflected by the slowness of the reaction leading to the desired product (several days at room temperature). Many coordination/decoordination steps have to occur before the system finds its thermodynamic well. The desired [2]pseudorotaxane was formed under thermodynamic equilibrium and, thus, it was obtained pure as a deep-red crystalline solid without further purification. X-ray quality crystals were grown from acetone/diethyl ether by diffusion. The crystallographic structure of the dicopper(I) [2]rotaxane dimer is represented in Figure 15. The most striking feature of the structure is its linear extended arrangement, which results in a distance between the two terminal phenolic oxygen atoms of 36.3 Å. The two copper atoms are 18.3 Å apart and occupy similar environments. The coordination tetrahedron around both copper(I) atoms are strongly distorted.

CuI-induced double threading process leading to a dicopper(I) rotaxane dimer. The reaction is quantitative. The structure of the doubly threaded compound obtained is shown in Figure 15.

X-ray structure of the dicopper(I) rotaxane dimer. The pale gray ring-and-string component and the black subunit are identical. The carbon atoms are pale gray or black. The two copper(I) centers are green. The nitrogen and oxygen atoms are deep blue and red, respectively.
In order to complete the synthesis of the switchable dimer rotaxane we had to introduce additional function. In particular, the project involved two potential binding modes to a transition-metal center for the organic backbone: four-coordinate or five-coordinate. Tridentate ligands of the terpy family were, thus, introduced simultaneously to the stoppers, whose function was simply to prevent unthreading of the molecular filament from the rings they were threaded through. The last organic reaction of the synthesis sequence is shown in Figure 16. The functionalized terpy represented in the upper part of Figure 16 incorporates a tridentate chelating group and is attached to a bulky stopper. It was prepared separately and subsequently it was attached to both ends of the dicopper(I) precursor of Figure 15. This last reaction afforded the muscle-like compound depicted in Figure 16 (bottom) as a dark-red solid in 60 % yield. The last coupling step not only allowed the introduction of terdentate sites on both sides of the molecular assembly but also afforded a real rotaxane which can not undergo any unthreading reaction.

Synthesis of the complete “muscle” in its extended situation. Functionalization of the [2]pseudorotaxane dimer, precursor to the “muscle”, by attaching a terpy-bulky stopper conjugate to its both ends. The “muscle” represented at the bottom of the Figure is in its extended situation.
As usual in our group, CuI was used as the gathering and templating metal. The movement was induced by a chemical reaction, namely a metal exchange. As shown in Figure 17 in a very schematic fashion, the doubly threaded compound can interact simultaneously with two metal centers, either in a four- or in a five-coordinate geometry each.

Contraction/extension of a [2]rotaxane dimer by gliding one strand along the other one. The W-shaped fragment represents a terdentate chelating group such as terpy, whereas the U-shaped species is a bidentate ligand of the dpp type.
Due to the synthesis procedure, each CuI center was coordinated to two 1,10-phenanthroline units, resulting in a four-coordinate situation. This binding mode corresponds to an extended geometry (upper part of Figure 17), whereas coordination of a divalent metal ion such as Zn2+ affords a five-coordinate situation corresponding to interaction of each metal center with one 1,10-phenanthroline or dpp unit and one terpy. It corresponds to a contracted geometry (bottom of Figure 17).
As shown in Figure 18, by treating the dicopper(I) pseudorotaxane dimer with KCN, the metal-free ligand was obtained in quantitative yield (not shown on the Figure). After remetalation using Zn(NO3)2, the colorless dizinc complex (bottom) was obtained quantitatively. It is now in the contracted form. The extended dicopper(I) dimer could be regenerated by the reaction of the dizinc complex with an excess of [Cu(CH3CN)4]PF6 at room temperature. Using a metal-exchange reaction, the muscle-like compound is thus set in motion. The CuI complex corresponds to the extended form and the ZnII-complexed species is the contracted state. A metal-exchange reaction was utilized to set the muscle in motion due to the fact that the electrochemical method turned out to be inefficient. The copper(I) complex was oxidized with the purpose of contracting the system to a dicopper(II) complex with two five-coordinate CuII centers but the motion was very slow, which led us to use a chemical signal. The kinetic barrier to the rearrangement is very large due to the fact that the gliding motion of a given thread is strongly coupled to that of the other thread. The tight connection between the two subunits makes it impossible for the [2]rotaxane dimer to move in a stepwise manner.

The two forms of the muscle-like [2]rotaxane dimer. Whereas the interconversion between both forms is easily induced by metal exchange, oxidation of copper(I) to copper(II) (upper form in the Figure) was unfortunately not followed by rearrangement of the complex to the extended situation, at least on a reasonable timescale. Due to the rigid structure of the molecule, the gliding motions of both threads along one another have to take place simultaneously and in a concerted manner. The kinetic barrier to the global motion is, thus, very high, which explains why the motion is so slow when no demetalation step is involved.
The two rotaxane dimers of Figure 18 represent one of the first examples of a unimolecular linear array able to elongate and contract at will under the action of a given stimulus. From a Corey–Pauling–Koltun (CPK) model, one can estimate that the length of the compound changes from 83 to 65 Å between both situations (Figure 18). This corresponds roughly to the same relative amount as what is found in natural muscles (ca. 27 %).
It is noteworthy that several muscle-like compounds have been proposed in recent years. In most cases, they were set in motion using principles which were somewhat different from ours. It is not the place in this article to review in detail the work done by other groups in this area, but we would like to mention some of the most significant contributions. Several groups prepared dynamic [2]rotaxane dimers based on cyclodextrins which, under the action of a given signal, could be contracted or elongated.39, 40 Reversible molecular motions mimicking natural muscles proceed through hydrophobic forces, size constraints, solvent polarity changes, or photochemical isomerization of azobenzene or stilbene derivatives.
Another family of muscle-like compounds was developed based on other types of interlocking architectures. These systems contain two types of recognitions sites, one of them being based on hydrogen bonding (and thus pH-sensitive) and the other one operating via acceptor–donor interactions. A few years ago, Stoddart and co-workers showed that a contraction and expansion motion was triggered by changing the pH of the solution containing the rotaxane.41 They described a new bistable rotaxane dimer architecture in 2008, namely a two-component [c2]daisy-chain topology. This system consists of two self-complementary interlocking molecular fragments in which a secondary ammonium-ion-containing lateral arm is attached to a crown ether. The end of each arm is connected to a bulky stopper. Each crown ether glides between the two specific binding sites incorporated in the thread, the motion being triggered by modifying the pH. The gliding motions result in contracting or elongating the rotaxane dimer.
Simultaneously to the former piece of work, Coutrot et al. published another example of a pH-switchable molecular muscle42 based on a related principle. The system incorporates a cyclic rotaxane dimer consisting of two interlocking monomers. Again, each monomer incorporates a crown ether attached to a linear fragment. This fragment contains two different stations and the system can be switched from a contracted state to an elongated situation by modifying the pH. A remarkable extension of Coutrot's work was published in 2012.43 Giuseppone and co-workers designed and synthesized a pH-driven muscle-like system similar to Coutrot's “muscle” in which [2]rotaxane dimers are linked together linearly by taking advantage of a very efficient coordination-chemistry-based polymerization process. A remarkably high degree of polymerization was obtained (ca. 3000 units) for both the contracted and extended forms. As in Coutrot's work, at low pH, a doubly threaded dimer adopts an extended geometry. At basic pH, the compound adopts a contracted form. These two states were very convincingly characterized by dynamic light-scattering and small-angle neutron scattering experiments. This remarkable piece of work is probably one of the first examples of molecular muscle operating at a microscopic scale and, thus, functioning as a real device.
Conclusions
The main field of research of our group since the beginning of the 1980s has clearly been that of transition-metal chemistry. In relation to the molecular machine area, it should be stressed that the metals had a dual function:
-
Interlocking and knotted ring compounds were obtained by using transition metals such as CuI in particular for gathering and disposing in a precisely defined geometry various organic fragments before they were incorporated in the target molecule.
-
The presence of transition metals provided the molecular systems with electrochemical or photochemical properties, which allowed to set given fragments of the molecule in motion and thus to obtain molecular machines.
Generally speaking, motivation for doing research in the field of controlled dynamic systems of the muscle-like family is manifold. The most important one is related to the scientific challenge that the synthesis and the study of complex and functional molecular systems that can be set in motion in a controlled way represents. Making contractile and extensible molecules of the rotaxane dimer type was probably not even envisioned a few decades ago due to expected synthetic and analytical difficulties. Preparing and studying linear or rotary motors or artificial muscles is related to the fascination that biology exerts on chemists and engineers. It is very challenging for a synthetic chemist to make a molecule or molecular system whose function and mode of action are reminiscent of those of biological systems. The few examples discussed in the present chapter are certainly very remote from natural muscles and they are undoubtedly very primitive compared to their natural analogues. Nevertheless, it was a great challenge to elaborate these compounds and to show that they can be set in motion in a well-controlled fashion.
The field of molecular machines in general, still a basic science area of research, has been acknowledged by the Royal Swedish Academy of Science and the Nobel Committee. I would like to thank these prestigious organizations for this recognition. This field is nowadays more than 20 years old and it is certainly time to address the question of applications, either in a general fashion for machine-like compounds or more precisely for contractile and extensible molecular species. In which field of application will the area of molecular muscles be the most important in the future? It is today very risky to give an answer to this question. Presently, one of the most impressive extensions of molecular machines towards applications is concerned with “molecular computing” and the molecular approach to electronic memory devices. The groups of Stoddart and Heath reported promising work in this area.44
Many types of nanodevices and nanomachines can be envisaged in relation to chemical applications (for example, sorting and transport of molecules in solution or through membranes) but also to purely mechanical applications. In the future, nano- and microrobots able to fulfil a large variety of functions (from medicine to everyday life) will have to be articulated. The use of molecular components to control their movements is certainly a promising possibility. In a relatively long-term prospective, artificial muscles of various lengths (from microns to millimeters or even centimeters) might be needed for various applications such as humanoid robots, actuators for microfluidic science and technology, or prosthetic organs. However, in spite of all the possibilities offered by molecular machines in terms of potential applications for the future, it should be stressed that basic research is still of utmost importance and is, or has been, at the origin of the many technologies which are nowadays part of our daily life.
Finally, I would like to stress that the recent book written by Bruns and Stoddart represents a monumental piece of work which, today, gathers all the important data obtained by hundreds if not thousands of researchers since the beginning of catenanes, rotaxanes, and molecular machines. Any interested reader must, of course, have a copy of this book.
Acknowledgements
The work on molecular machines performed in Strasbourg was financially supported by the University of Strasbourg (formally Université Louis Pasteur), the CNRS (Centre National de la Recherche Scientifique), the European Commission, and the International Center for Frontier Research in Chemistry (ICFRC). We would like to thank these organizations. I also acknowledge the important contribution of Dr. Christiane O. Dietrich-Buchecker (deceased in 2008). Her contribution to the early work performed in Strasbourg on molecular catenanes and molecular machine prototypes has really been crucial. I would also like to thank my mentor, Prof. Jean-Marie Lehn. Jean-Marie has represented a model for me since I worked with him as a PhD student. I am grateful to two teachers who had a strong influence on me: Prof. Guy Ourisson (organic chemistry) and Raymond Weiss (inorganic and physical chemistry). I would also like to express my gratitude to Prof. Malcolm L. H. Green. As a postdoctoral fellow in his team, I learnt transition-metal and organometallic chemistry, which also had a strong impact on the future projects of my group. Many researchers have been associated to the work of our team, “Laboratoire de Chimie Organo-Minérale”. In spite of the expected and unexpected difficulties they frequently had to face in the course of their work, these researchers have always been very enthusiastic and efficient. I owe them the success of our research team. The names of our CNRS researchers or University members (from assistant to full professors), of our PhD students and postdoctoral researchers as well as those of other visitors who contributed to our research are listed in Figures 19 and 20. Finally, I thank my parents, my wife Carmen, and my son Julien, for their continuous support and for their forgiveness regarding my pace of work and travel.

1. Permanent researchers of the “Laboratoire de Chimie Organo-Minérale” (LCOM): University members and CNRS researchers including the late Dr. Christiane Dietrich-Buchecker (1942–2008) and late Prof. Jean-Marc Kern (1944–2004). 2. PhD students listed by chronological order. The two first students, Pascal Marnot and Romain Ruppert, started their PhD work in 1980.

Postdoctoral researchers and other visitors.
Biography
I was born on October 21st, 1944 in Paris, just after the end of the second world war and a few months after Paris had been liberated by the allies and the French army led by General Charles de Gaulle (19–25 August 1944). My mother's name was Lydie Angèle Arcelin and her family came mostly from Normandy. She was born in 1920. My father was Camille André Sauvage and came from the Northern part of France. Camille Sauvage was known as a successful jazz musician, both conductor and clarinetist on top of being a composer. Just after the war, he was a popular jazz player and, later on, he composed for French radio, television, and movies. When I was a baby, my parents broke up. I stayed with my mother, while my father departed to pursue an artist's life. While I was still a young child, my mother met an officer in the air force, Marcel Louis Grosse, and they founded a family. I was, thus, fortunate to have a stepfather who took care of me until I became an adult and whose role was truly that of my father. Thus, I always considered him as my real father and I still do. Since my stepfather was in the military and because my parents loved to travel from one place to another, I had a very mobile childhood. When I was three years old, we moved to North Africa, as Algeria and Tunisia were still French colonies at that time. I still have vivid memories of our time in Zarzouna, a small village close to Bizerte in Tunisia. I also spent some time near Oran, in Algeria. I went to school in Tunisia between the ages of 5 and 7. At that time, the French kids and the local Tunisian kids were together in the same classes and I do not believe there were any difficulties related to the fact that the French kids and the Tunisian ones were mingling.
My mother and my grandmother, Suzanne Arcelin, were very close to one another, and my grandmother, who used to live in Paris, visited our family a few times. On a particular occasion, we went to southern Tunisia for sightseeing and the photo which is shown below is particularly representative. It was taken when I was about 21 years old.

In Tunisia with my mother and, on the left, my grandmother.
From 1951 to 1952, my family spent some time in the USA since my stepfather had to become a military engineer in the field of radar applications, a relatively new technology at the beginning of the 1950s. We thus spent 6 months in Saint Louis, Missouri, followed by an additional period of 22 months in Denver, Colorado. I had no difficulty in adapting and in Denver, in particular, I used to play with the other children of our neighborhood. In Saint Louis, we used to go to the movie theater from time to time and it was, of course, a very enjoyable event for me. The picture shown below is interesting in the sense that one can see who were the most popular actors of the time.

After a movie in Saint Louis (USA) in 1951.
When we returned to France, we started a long period of itinerancy, spending a few months in a given place before moving to a new city. I thus went to 4 or 5 different schools in the west part of France and in the Paris area when I was 8 to 10 years old. My mother became ill when I was about 10 and it was a difficult moment for my family. She had contracted tuberculosis, which was a very serious disease in the 1950s. I was thus mostly with my grandmother for about a year, in the family village of Pacy-sur-Eure in Normandy. After this period, I was again with my parents, who moved to the eastern part of France in the Lorraine region. When I was 15 years old we moved to a small village in the north of Alsace, Drachenbronn, because my stepfather had been transferred to the radar station located on the Maginot Line named “Base Aérienne 901 Drachenbronn”. This move coincided with the beginning of my high school studies and I thus went to the high school of Haguenau, a middle-size city not far from air base 901. I started to be very interested in chemistry when I was 15 or 16 years old and, in particular, I liked to play with natural molecules such as chlorophylls which I extracted from plants. I had a small and very primitive chemistry lab in the cellar where I was separating chlorophylls on paper or distilling various mixtures. At school, I was probably better in mathematics than in physics or chemistry, but the interest of pupils for various topics is obviously very dependent on the personality of the teacher.
After my “baccalauréat” (the French examination obtained at the age of 18 which enables to go to university), I decided to enter a special and highly competitive structure named “classes préparatoires”, which was aimed at preparing the young people to compete for admission in engineering schools. Thus, after two years of a rigorous regime where leisure was limited to a strict minimum, I succeeded and I was admitted to the Chemical Engineering School of Strasbourg. This was exactly what I wanted, since I could stay in my new, but already beloved, city. I obtained my engineer diploma in 1967 and I started my PhD thesis in 1967 under the guidance of Prof. Jean-Marie Lehn. Jean-Marie had founded his own research team a few years before I started to work with him. He was only 5 years older than I and his research team was developing rapidly in terms of size and breadth of his research interests. Being more a physical chemist up until 1967, he had become interested in making new molecules with novel properties when I started with him. At the time, Bernard Dietrich, formally a technician, decided to start his studies so as to become a graduate student, hoping also to do a PhD thesis. Bernard rapidly became my best friend and we worked together in the most friendly atmosphere imaginable between 1968 and 1971. Under the guidance of Jean-Marie, we were highly successful since we made the first macrobicyclic compounds able to encapsulate various ions, including alkali and alkaline earth metal cations (cryptands and cryptates for the metal-free compounds and their complexes, respectively). In Jean-Marie Lehn's research group, I was able to acquire a solid background in organic and physical chemistry, thanks to my own work and to various seminars and group meetings as well as to the many hours I used to spend in the library. Discussions with Prof. Lehn and with other members of the group were also very fruitful. Equally important was the influence that Jean-Marie had on me in terms of the relationship between researchers within a group and, in particular, I enjoyed his way of managing a research team. He was very direct, placing basically no barrier between him and the PhD students or postdocs he was working with. In other words, hierarchy was reduced to its minimum and this is something which I tried to preserve later on in my own group. Inspired by Jean-Marie's passion for science, I also became enthusiastic and determined to devote my life to research.
I met my wife Carmen in 1967 and we got married in February 23. Carmen was a student in History of Art and Archaeology. She was particularly interested in ancient ceramics and had participated in several excavation campaigns on the Anatolian plateau, in Turkey. Although we were not especially religious, we had a religious wedding, mostly to respect family traditions. The wedding took place in Thierenbach, a village in the south of Alsace which used to be a place of pilgrimage and which is nowadays famous for its basilica.

The wedding ceremony in Thierenbach, February 8, 1971.
We were very happy to become parents of a baby boy, Julien Clément Sauvage, on July 13, 1975. It was a great joy for us. Since Julien's prime childhood, we have been very close to him. In 2011, Julien married Diana, originally from Columbia, and since 2012 they have settled down in San Francisco. They had a baby on April 9, 2016 so that Carmen and I became the happiest grandparents in the world.
At the end of my PhD thesis I obtained a CNRS position as “chargé de recherche” (research assistant) in Jean-Marie Lehn's group, which corresponded exactly to what I was so eager to get.
After my PhD thesis, I obtained a postdoctoral fellowship in Oxford (UK), where I spent a year in the research group of a very visible organometallic chemist, Dr. Malcolm L. H. Green. Malcolm was considered as one of the most brilliant former students of Prof. Geoffrey Wilkinson (1973 Nobel Laureate in chemistry with Prof. Ernst Otto Fischer). He was a very influential person in expanding my interests to transition-metal chemistry and organometallic chemistry. He was also a friendly person, who used to do experimental work by himself from time to time. Life in Oxford was particularly pleasant and we used to enjoy the city, its colleges, and its parks. We easily adopted the way of life of the other members of the Oxford community.
After my postdoctoral stay in Oxford, I came back to Strasbourg and, more precisely, to the Lehn laboratory as a permanent researcher. After some work in the field of chiral crown ethers, we initiated, Jean-Marie and I, a research project in a new field, at least in Strasbourg. It was related to photochemistry and solar energy. The first oil crisis took place in 1973 and it was a clear signal that alternative energies had to be found and that sustainable energies were crucially needed. Solar energy was an obvious and especially attractive option. A particularly appealing project was that of splitting the water molecule to H2 and 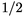 O2 in order to generate a nonpolluting fuel, H2, using photonic energy. Such a big project had already been explored for many years by several groups and there were already discussions and research works published on this general topic in the 1950s and later on. More or less at the same time, the ruthenium complex Ru(bipy)32+ (bipy=2,2′-bipyridine) was shown to display promising electronic properties in its ground or excited states, in particular in relation to electron transfer and potential photochemical water splitting. In 1977 we published one of the very first systems leading to photochemical water reduction to H2 based on a combination of species such as, in particular, Ru(bipy)32+ as a photoactive species and Rh(bipy)33+ as an electron relay leading to H2 formation. After two years of studies on this original system (with Jean-Marie Lehn and Michele Kirch) and related ones, as well as on the development of a light-driven oxygen-generating system from water (with Jean-Marie Lehn and Raymond Ziessel), I was lucky enough to be promoted to the position of CNRS Research Director, the equivalent of University Professor. I thus founded my own research group in 1980, at first with two highly motivated PhD students, Pascal Marnot and Romain Ruppert. After one or two years, Jean-Paul Collin, a CNRS fellow, and Marc Beley, an Associate Professor, joined our small team. Simultaneously, a good friend of mine, Christiane Dietrich-Buchecker, also joined. As it is often the case for young research teams, we tackled several research projects in parallel in relatively remote areas. The electrochemical reduction of CO2 using [Ni-cyclam]2+ as electrocatalyst led to remarkable data, since CO2 could be reduced very selectively to CO in water. This was somewhat surprising, since H2 was expected to be obtained as a major reduction product. We also did work in homogeneous catalysis and inorganic photochemistry. In this latter field, our projects were mostly triggered by a collaboration with David R. M. McMillin who was on sabbatical leave in Strasbourg. David was an already well-established photochemist and photophysicist. He was a professor at the university of Purdue (West Lafayette, USA) and his main field of research was copper(I) complexes photochemistry. This collaboration between our group, with its skill in organic synthesis, and David led to a series of particularly interesting photoactive copper complexes. Perhaps even more important, it led to a copper(I) complex containing two intertwined organic ligands which appeared to be the ideal precursors to a compound comprising two interlocking rings. It was thus very tempting to jump from inorganic photochemistry to interlocking ring compounds which, at the beginning of the 1980s, seemed to be practically inaccessible molecules. This jump was made possible due to the expertize of Christiane Dietrich-Buchecker, who was a great organic chemist. After a few discussions within our team, we decided to take the risk and to embark on a totally new project concerned with the synthesis of catenanes (i.e. interlocking ring compounds). Within a few months, Christiane was able to develop an efficient preparative procedure for making our first [2]catenane (containing 2 interlocking rings). Respectable quantities could be obtained: batches of 0.5 g could be prepared, in particular by Jean Weiss, a PhD student also supervised by Christiane, and the first compound was fully characterized using a variety of techniques. 1H NMR provided the first convincing evidence that a [2]catenane was indeed produced. These experiments were carried out by Jean-Pierre Kintzinger, a friend of mine and the brother-in-law of Christiane, who was at the same time an NMR expert. We published our first paper in this field in 1983 in an acceptable, but not high-impact, journal (Tetrahedron Letters). It was for us the beginning of a new era, mostly but not exclusively devoted to interlocking rings compounds and knotted molecules. The field has often been referred to as “Chemical Topology” due to the fact that the compounds have nonplanar molecular graphs. In other words, contrary to almost all the molecules known, it is impossible to draw them in a plane (i.e. on a sheet of paper) without crossings, regardless of the deformation the molecule can be subjected to.
O2 in order to generate a nonpolluting fuel, H2, using photonic energy. Such a big project had already been explored for many years by several groups and there were already discussions and research works published on this general topic in the 1950s and later on. More or less at the same time, the ruthenium complex Ru(bipy)32+ (bipy=2,2′-bipyridine) was shown to display promising electronic properties in its ground or excited states, in particular in relation to electron transfer and potential photochemical water splitting. In 1977 we published one of the very first systems leading to photochemical water reduction to H2 based on a combination of species such as, in particular, Ru(bipy)32+ as a photoactive species and Rh(bipy)33+ as an electron relay leading to H2 formation. After two years of studies on this original system (with Jean-Marie Lehn and Michele Kirch) and related ones, as well as on the development of a light-driven oxygen-generating system from water (with Jean-Marie Lehn and Raymond Ziessel), I was lucky enough to be promoted to the position of CNRS Research Director, the equivalent of University Professor. I thus founded my own research group in 1980, at first with two highly motivated PhD students, Pascal Marnot and Romain Ruppert. After one or two years, Jean-Paul Collin, a CNRS fellow, and Marc Beley, an Associate Professor, joined our small team. Simultaneously, a good friend of mine, Christiane Dietrich-Buchecker, also joined. As it is often the case for young research teams, we tackled several research projects in parallel in relatively remote areas. The electrochemical reduction of CO2 using [Ni-cyclam]2+ as electrocatalyst led to remarkable data, since CO2 could be reduced very selectively to CO in water. This was somewhat surprising, since H2 was expected to be obtained as a major reduction product. We also did work in homogeneous catalysis and inorganic photochemistry. In this latter field, our projects were mostly triggered by a collaboration with David R. M. McMillin who was on sabbatical leave in Strasbourg. David was an already well-established photochemist and photophysicist. He was a professor at the university of Purdue (West Lafayette, USA) and his main field of research was copper(I) complexes photochemistry. This collaboration between our group, with its skill in organic synthesis, and David led to a series of particularly interesting photoactive copper complexes. Perhaps even more important, it led to a copper(I) complex containing two intertwined organic ligands which appeared to be the ideal precursors to a compound comprising two interlocking rings. It was thus very tempting to jump from inorganic photochemistry to interlocking ring compounds which, at the beginning of the 1980s, seemed to be practically inaccessible molecules. This jump was made possible due to the expertize of Christiane Dietrich-Buchecker, who was a great organic chemist. After a few discussions within our team, we decided to take the risk and to embark on a totally new project concerned with the synthesis of catenanes (i.e. interlocking ring compounds). Within a few months, Christiane was able to develop an efficient preparative procedure for making our first [2]catenane (containing 2 interlocking rings). Respectable quantities could be obtained: batches of 0.5 g could be prepared, in particular by Jean Weiss, a PhD student also supervised by Christiane, and the first compound was fully characterized using a variety of techniques. 1H NMR provided the first convincing evidence that a [2]catenane was indeed produced. These experiments were carried out by Jean-Pierre Kintzinger, a friend of mine and the brother-in-law of Christiane, who was at the same time an NMR expert. We published our first paper in this field in 1983 in an acceptable, but not high-impact, journal (Tetrahedron Letters). It was for us the beginning of a new era, mostly but not exclusively devoted to interlocking rings compounds and knotted molecules. The field has often been referred to as “Chemical Topology” due to the fact that the compounds have nonplanar molecular graphs. In other words, contrary to almost all the molecules known, it is impossible to draw them in a plane (i.e. on a sheet of paper) without crossings, regardless of the deformation the molecule can be subjected to.
Besides chemical topology and molecular machines, our group has been active in various relatively remote fields. The principal alternative research area has been that of artificial photosynthesis, with a particular emphasis on photoinduced charge separation, one of the key processes of natural or artificial photosynthesis. In order to elaborate efficient models of the natural photosynthetic systems, and in view of realizing the complete water splitting cycle in the future, our group synthesized numerous multicomponent complexes, either incorporating metal-complexed porphyrins or second- or third-row transition-metal complexes (Ru, Os, Rh, and Ir) able to undergo light-induced charge separation. Following the synthetic work, the photochemical and photophysical properties of most of the compounds were investigated in various places by more physical chemistry oriented research teams than ours. In particular, a long-term collaboration with renowned photochemists located in Bologna turned out to be specially pleasant and fruitful (Balzani and his co-workers, University of Bologna, or Flamigni and Barigelletti, Consiglio Nazionale delle Ricerche, Bologna). Some of the charge-separated states were shown to be remarkably long-lived, thus paving the way to real artificial photosynthetic devices reminiscent of the photosynthetic apparatus of green plants or photosynthetic bacteria.
I would like to stress that encounters with various people played a very important role in my professional life. Two teachers were particularly influential when I was a student: Raymond Weiss, who was a very rigorous physical and inorganic chemist, and Guy Ourisson, an exceptional organic chemist who was able to convince all the students he was teaching to that organic chemistry is exciting and can even be fun. Jean-Marie Lehn had also a great impact on my enthusiasm for science, and to me he was the perfect model, although this model was totally out of reach. One of the most important encounters was that with Christiane Dietrich-Buchecker, a wonderful person and a great organic chemist, whose contribution to the scientific production of our group turned out to be determinant. Finally, it may appear as surprising that with Fraser Stoddart, we never looked at each other as competitors. Even more, we both tried to avoid any overlapping with the activities of the other research team. This is mostly due to the fact that we became friends at the end of the 1970s and this was the beginning of a faithful friendship which allowed us to work in a more serene atmosphere than if we had tried to overtake the other. Between 2010 and 2013, I was appointed as visiting professor at Northwestern University, where I collaborated primarily with Fraser and his team. I also enjoyed interacting with other colleagues in this great university. I am particularly grateful to Fraser for arranging for me to get such a position.
I would like to conclude with two pictures separated by approximately 29 years. The first one, Figure 24, was taken at the ceremony to honor Jean-Marie Lehn's Nobel Prize in 1987. This ceremony took place in the Great Lecture Hall of the Chemistry Department of our university. The second picture, Figure 25 is much more recent, since it was taken in Stockholm just after the 2016, December 10 ceremony.

One of the ceremonies held in Strasbourg (1988) to honor Jean-Marie Lehn's Nobel Prize (1987). On this special occasion, the chemistry community gathered in a friendly atmosphere (as testified by the number of smiley people in the picture). From left to right, the four persons at the front are Jean-Marie Lehn, Jean-François Biellman, myself, and Guy Ourisson. My very good friend Bernard Dietrich (1940–2004), who was also one of the main contributors to the early work on cryptands and cryptates, is in the second row just behind J.-F. Biellman.

Picture taken on the stage, after the Nobel Ceremony with the Royal Family. From left to right : Annelie Almkvist, our Attachée, Diana Sistiva, our daughter in law, Julien Sauvage, our son, Carmen Sauvage, my wife, myself, and Jean-Marie Lehn.

