

Rapid Access to an Enantioenriched Spiro Bisindole Directly from Indole and Acetone
Abstract
Disclosed here is a “one-pot self-assembly” strategy for the enantioselective synthesis of a spiro bisindole molecule directly from indole and acetone. This convenient synthesis contrasts drastically to the typical long sequence required for other privileged C2-symmetric spirocycles. The use of an alcohol additive in cooperation with chiral phosphoric acid (CPA) catalysis proved crucial to the good efficiency and enantioselectivity. The favorable interaction between the additive and CPA was confirmed by nuclear magnetic resonance (NMR) and density functional theory (DFT) studies. The obtained enantiopure C2-symmetric bisindole was demonstrated as a useful backbone of chiral catalysts/ligands.
Modern asymmetric synthesis has benefitted significantly from the development of novel chiral catalysts.[1-4] Specifically, C2-symmetric spirocyclic frameworks, such as 1,1′-spirobiindane-7,7′-diol (SPINOL), represent one of the most prominent examples of privileged backbones for diverse chiral catalysts.[5-8] Their rigid spirocyclic skeletons combined with the flexibility to tune steric environment have enabled their extraordinary performance in catalysis. The success of SPINOL has also stimulated extensive efforts in the design of diverse analogues.[9-20] For example, heteroatoms or additional rings have been incorporated into the two spiro five-membered rings. More recently, as reported by us and the Tan laboratory, the phenol rings in SPINOL have also been replaced by naphthol and indole rings, leading to 2,2′,3,3′-tetrahydro-1,1′-spirobi[phenalene]-9,9′-diol (SPHENOL) and a spiro bisindole (SPINDOLE-1).[21-25] Their synthesis was simplified by facile enantioselective spirocyclization owing to the increased nucleophilicity and well-defined nucleophilic site of the naphthol and indole rings (Scheme 1a).

While the above chiral spirocyclic structures have been demonstrated as useful backbones of chiral catalysts in various reactions, it is important to note that their syntheses have remained far from ideal. The most general approach is the controlled spirocyclization of a pre-synthesized ketone bearing two internal electron-rich arenes by way of two sequential Friedel–Crafts reactions.[5-8, 24, 25] The ketone precursor typically requires a few steps to synthesize, featuring an aldol condensation from acetone and a suitable aldehyde that is often not commercially available. Sometimes additional operations are required in the sequence, such as protection/deprotection, blocking/de-blocking, and chiral resolution. Consequently, these intriguing spirocyclic catalysts have been somewhat overshadowed for large-scale applications.
In this context, the design of an analogue that can be easily accessed from readily available cheap materials with minimal synthetic operations would be ideal. Herein, we report a “one-pot self-assembly” strategy to access a chiral spiro bisindole (SPINDOLE-2) directly from indole and acetone (Scheme 1b).[26-29]
The reactions between indole and acetone can potentially generate a mixture of various products due to the multiple nucleophilic sites and rich reaction patterns of indole.[30-38] However, the enantioselective synthesis of a useful spiro bisindole from this reaction remains elusive. Chiral phosphoric acids (CPAs) were chosen as catalysts for our study in view of their outstanding performance in indole chemistry and related synthesis.[21-26, 28, 39-44] Unfortunately, with 2,2′-binaphthol (BINOL)-derived CPA A1, the reaction did not form any spirocyclic product at room temperature. Instead, bisindole 3 was the major product (Table 1, entry 1). However, the reaction at 80 °C did form a spiro structure, which was confirmed as bisindole 1 (entry 2). Next, we screened some other CPA catalysts aiming to improve the yield and enantioselectivity. The sterically bulky catalyst A2 could not promote the desired reaction (entry 3). Nevertheless, A3 and A4 both produced 1 as the major product, with the latter being more enantioselective (entries 4–5). The SPINOL-derived A5 bearing the same side arms did not give the desired product (entry 6). In these reactions, some other products, including 2, 3, and 4, accounted for the mass balance.
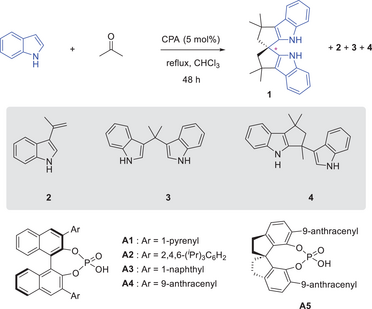 |
||||
| Entry | CPA | Solvent | Yield of 1(%) | ee (%) |
|---|---|---|---|---|
| 1 | (R)-A1 | CHCl3 | <5b) | − |
| 2 | (R)-A1 | CHCl3 | 27 | 15 |
| 3 | (R)-A2 | CHCl3 | <5 | − |
| 4 | (R)-A3 | CHCl3 | 57 | 51 |
| 5 | (R)-A4 | CHCl3 | 52 | 58 |
| 6 | (R)-A5 | CHCl3 | <5 | – |
| 7 | (R)-A4 | Acetone | 20 | 21 |
| 8 | (R)-A4 | EtOAc | <5 | – |
| 9 | (R)-A4 | Toluene | 42 | 14 |
| 10 | (R)-A4 | CDCl3 | 65 | 13 |
- a) Reaction conditions: indole (0.2 mmol), acetone (1.0 mmol), CPA (0.01 mmol), solvent (1.0 mL), 80 °C (heating plate temperature), and 48 h. Yield was based on 1H NMR analysis of the crude reaction mixture. The ee was determined by chiral HPLC.
- b) Run at RT. Compounds 3 (66%) and 4 (33%) accounted for the mass balance.
With the best catalyst A4, we evaluated other solvents, but with no improvement in enantioselectivity (entries 7–10). Nevertheless, it was found that CDCl3 led to a substantial decrease in enantioselectivity (13% ee), which should not be simply due to the isotope effect. We suspected that the trace amount of EtOH present in commercial CHCl3 (but not in CDCl3) made this difference.
Then we employed freshly distilled CHCl3 for this reaction (Table 2, entry 1). Intriguingly, the enantioselectivity dropped to 13% ee, consistent with that from CDCl3. EtOH was then added on purpose, which led to obvious improvement in enantioselectivity (entry 2). The positive effect of EtOH prompted us to further evaluate other alcohol additives (entries 3–7). While some of them could improve the yield but most led to decreased enantioselectivity. Among them, 3,5-bis(trifluoromethyl)benzyl alcohol B1 provided improvements in both yield and enantioselectivity (entry 6). Other hydrogen-bond donors such as thiourea B3 and its chiral analogues, were not able to improve the results (entry 8). Increasing the loading of B1 could not further improve the outcome. We also examined the effect of drying agents, considering water was generated. Among them, MgSO4 led to slight improvement (entries 9–11). Finally, decreasing the temperature could lead to further improvement in enantioselectivity, but with slight decrease in yield (entry 12).
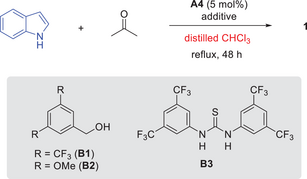 |
|||
| Entry | Additive | Yield (%) | ee (%) |
|---|---|---|---|
| 1 | – | 61 | 13 |
| 2b) | EtOH | 54 | 33 |
| 3 | BnOH | 58 | 46 |
| 4 | PhOH | 68 | 28 |
| 5 | HFIP | 82 | 20 |
| 6 | B1 | 82 | 61 |
| 7 | B2 | 58 | 46 |
| 8 | B3 | 61 | 10 |
| 9c) | B1 | 45 | 72 |
| 10d) | B1 | 79 | 64 |
| 11e) | B1 | 88 | 64 |
| 12e), f) | B1 | 70 | 80 |
- a) Reaction conditions: indole (0.2 mmol), acetone (1.0 mmol), CPA (0.01 mmol), solvent (1.0 mL), additive (0.1 mmol), 80 °C (heating plate temperature), and 48 h. Yield was based on 1H NMR analysis of the crude reaction mixture. The ee was determined by chiral HPLC.
- b) EtOH (0.17 mmol) was added.
- c) 5 Å MS (50 mg) was added.
- d) Na2SO4 (50 mg) was added.
- e) MgSO4 (50 mg) was added.
- f) Run at 60 °C.
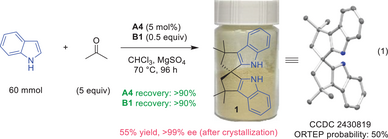
Since both indole and acetone are dirt cheap and the spiro bisindole product is crystalline, we envisioned that the current protocol might be sufficient for large scale synthesis. Thus, a 60 mmol reaction was run at 70 °C under the optimized conditions (Equation 1). After simple crystallization, the enantiopure product 1 was obtained as a yellow fine powder in 55% overall yield. Its structure was confirmed unambiguously by X-ray crystallography. Notably, both A4 and B1 could be fully recovered.
We also evaluated various substituted indoles with different electronic properties. In general, electronic-donating substituents provided the desired products with good efficiency and moderate to good enantioselectivity. However, electron-withdrawing substituents dramatically affected the reaction efficiency. Cyclohexanone was also tested in place of acetone. Unfortunately, no desired spiro bisindole was obtained, likely due to the increased steric hindrance in the C─C bond-forming events (more details in the Supporting Information).
The N─H motifs in the newly generated enantiopure bisindole 1 could be potential handles to attach functional groups (Scheme 2). For example, sequential treatment of compound 1 with n-BuLi followed by 1-isothiocyanato-3,5-bis(trifluoromethyl)benzene generated chiral bis(thiourea) 5 in almost quantitative yield. Moreover, the diphenylphosphine and thiourea units could be sequentially installed to give 6. Similarly, a pyridine unit could be grafted together with a phosphine moiety in 7. The bis(phosphine) 8 was also easily accessible in 84% yield, which could generate Pd-complex 9 upon mixing with Pd(PhCN)2Cl2. Furthermore, partial reduction of 1 by NaCNBH3 formed secondary amine 10, a potential chiral amine organocatalyst. Moreover, two Bpin units could be installed selectively to the 7-position of the indole rings by iridium catalysis, leading to bis(borate) 11. Subsequent Pd-catalyzed cross-coupling with PhBr allowed facile introduction of two phenyl rings, thus illustrating the flexibility to tune the steric environment of this spiro bisindole skeleton when needed.

The above derivatives are potential chiral catalysts for asymmetric transformations. For example, the Pd-complex 9, together with an allyl Grignard reagent, served as an efficient pre-catalyst for the enantioconvergent allylic substitution between 13 and 14 with high efficiency and excellent enantioselectivity (Scheme 3a). Moreover, an enantioselective Pd-catalyzed allylic amination of 16 could be achieved with bis(phosphine) 8 as ligand (Scheme 3b). In cooperation with a rhodium catalyst, ligand 8 could induce excellent enantiocontrol in the efficient hydrogenation of enamide 19, leading to quantitative formation of the amino acid derivative 20 (Scheme 3c). Finally, the chiral secondary amine 10 could catalyze the enantioselective conjugate addition of N-methylpyrrole 21 to enal 22.[45] Upon in situ reduction, alcohol 23 was obtained in excellent enantiopurity (Scheme 3d). All these examples clearly illustrate the outstanding performance of our new spiro bisindole backbone.

A possible mechanism is proposed (Scheme 4).[30-36] The chiral acid first catalyzes the Friedel–Crafts reaction between indole and acetone. The resulting indolyl alcohol IM1 easily ionizes to carbocation IM2, which then deprotonates to form the experimentally observed olefin 2 and may be in equilibrium with imine IM3. Notably, IM2 is highly electrophilic and thus can react with various nucleophiles in the reaction system. For example, the reversible reaction with another indole molecule leads to bisindole 3, a compound initially observed and then gradually consumed during the reaction progress (see the Supporting Information for details). Alternatively, the reaction between IM2 and 2 affords indolyl carbocation IM4, which cyclizes at the C2-position of an indole motif to form 4, which was also experimentally observed. This step may involve the cyclobutane intermediate IM4-4 featuring an attack from the C3-position followed by a 1,2-alkyl migration. Next, a C─C bond cleavage is triggered by protonation of an indole motif (via IM5) to form IM6, with concomitant extrusion of an indole molecule. Further deprotonation leads to a key intermediate IM7. As a good nucleophile, it can further react with carbocation IM2 to form IM8, thus setting the stage for spirocyclization. This key step may involve a nucleophilic attack from the indole nucleophile to the 2-indole imine methide (2-IIM) motif.[46] The chiral acid serves as a bifunctional catalyst to activate both the 2-IIM and indole motifs, thereby generating the observed product 1 with enantiocontrol. Again, the spirocyclization may also involve initial attack at the C3-position (via IM8-1) followed by 1,2-alkyl migration.[47, 48] It is probably worth noting that the C3-to-C2 migration in this case may involve chemoselectivity issue since two σ-bonds in the cyclobutane ring can potentially migrate, but high selectivity was observed in this case.

Control experiments were performed to gain more insights into the mechanism (Figure 1a,i–iii). Compounds 2–4 all successfully generated 1 with comparable enantioselectivity when separately subjected to the standard reaction with acetone. The high enantioselectivity obtained from racemic intermediate 4 suggested that the product stereochemistry is determined by the chiral catalyst in a subsequent step. Under standard conditions, both N-methylindole and N-Boc-protected indole failed to afford the corresponding spiro bisindole products, thus consistent with the proposed essential role of the free N─H motif in the indole substrate for hydrogen bonding interaction of the catalyst (Figure 1a,v). These results agree with the proposed mechanism. The product ee values showed a linear relationship with the catalyst ee values (Figure 1b), thus consistent with the involvement of one CPA molecule in the enantiodetermining transition state.

We were intrigued by the dramatic influence of additive B1. When the protected methyl ether B1' was used, the reaction provided only 20% yield and 16% ee, in stark contrast to the standard reaction (Figure 1a,iv). Obviously, the free alcohol in B1 plays a critical role. To know more about the nature of this role, we further studied the possible interaction between B1 and A4. Upon mixing with A4, the proton nuclear magnetic resonance (1H NMR) signals of the two benzylic hydrogens of B1 showed significant up-field shifts. The shifts were greater with increased loadings of A4 (Figure 1c). Moreover, the original two enantiotopic hydrogens were split into two doublets, indicative of becoming diastereotopic, likely caused by interaction with a chiral molecule. Indeed, the 19F NMR signal of B1 also showed consistent shifts (see more details in the Supporting Information). We also observed obvious shifts of the 31P NMR signal of A4 when mixing with B1 (Figure 1d).[49] These results clearly indicate a favorable interaction between A4 and B1, probably via hydrogen bonding, which was confirmed by density functional theory (DFT) calculations (see the Supporting Information for details). We believe that such interactions may increase the acidity of the CPA catalyst and impose additional steric influence during asymmetric induction, thereby contributing to the observed enhanced catalytic performance.[50, 51]
To know more about the structure properties of the related spiro bisindoles, we compared some key parameters based on their X-ray crystallographic data (Figure 2). The dihedral angle (between the two indole rings) of SPINDOLE-2 is 90°,[52] which is in between those of SPINDOLE-1 (79.1°) and SPINDOLE-1-Ph2 (99.2°).[23] It has a much shorter N⋯N distance than the other two analogues. These distinctive features may allow SPINDOLE-2 to provide complementary performance when serving as a chiral catalyst backbone.

In conclusion, we have developed a “one-pot self-assembly” strategy for the rapid synthesis of a spiro bisindole and demonstrated its utility in asymmetric catalysis. The direct use of cheap materials, indole and acetone, in a simple operation constitutes the most prominent feature of this design, which contrasts drastically with the long synthetic sequence required for those conventional privileged C2-symmetric spirocyclic structures. The newly obtained bisindole has been validated as a useful chiral backbone of effective catalysts/ligands. Control experiments provided additional support for the mechanism, which involves many C─C bond formation and cleavage events. Despite the possibility of generating multiple products, the optimized protocol was able to achieve high efficiency and good enantioselectivity. The key to success lies in the use of a uniquely effective alcohol additive together with a suitable CPA catalyst. Their favorable interaction was confirmed by NMR and DFT studies. More detailed investigations are ongoing in our laboratory.
Acknowledgements
The authors thank the National Natural Science Foundation of China (22271242 and 22471232), the Hong Kong Research Grants Council (C6012-21G, 16309321, 16304322, 16309722, and 16310924), and Innovation and Technology Commission (ITC-CNERC14SC01) for financial support. The authors thank Dr. Herman H.Y. Sung for assistance in structure determination by X-ray crystallography.
Conflict of Interests
A provisional patent has been filed.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

