

Substratinspirierte Fragment-Fusion und -Erweiterung führt zu wirksamen LasB-Inhibitoren
Abstract
Extrazelluläre Virulenzfaktoren haben sich als attraktive Ziele in der aktuellen Antibiotikaresistenzkrise erwiesen. Der Gram-negative Erreger Pseudomonas aeruginosa sezerniert den Virulenzfaktor Elastase B (LasB), der eine wichtige Rolle im Infektionsprozess spielt. Hier berichten wir über einen submikromolaren, nicht-peptidischen, fragmentartigen Inhibitor von LasB, der durch sorgfältige visuelle Analyse von Strukturdaten entdeckt wurde. In Anlehnung an das natürliche LasB-Substrat wurde das ursprüngliche Fragment erfolgreich fusioniert und vergrößert. Der optimierte Inhibitor ist durch einfache Chemie zugänglich und behält seine Selektivität bei, wobei die Aktivität erheblich verbessert wird, was durch die Kristallstruktur von LasB im Komplex mit dem Inhibitor erklärt werden kann. Wir zeigen auch eine verbesserte in vivo-Wirksamkeit des optimierten Hits in Galleria mellonella-Larven, was die Bedeutung dieser Klasse von Verbindungen als vielversprechende Arzneimittelkandidaten unterstreicht.
Alternative Bindungsmodi werden häufig im Bereich des fragmentbasierten Wirkstoffdesigns beobachtet.1 Trotz des Potenzials, die Hit-to-Lead-Optimierung erheblich zu beschleunigen, gibt es nur wenige Beispiele für die erfolgreiche Verknüpfung/Fusion von Fragmenten oder die systematische Nutzung solcher unschätzbaren Quellen struktureller Informationen. Dies ist vermutlich auf eine Reihe von Bedingungen zurückzuführen, die erfüllt werden müssen, wie z. B. die Zusammensetzung des Linkers und die daraus resultierenden ADMET-Eigenschaften.2, 3 Wir vertreten die Auffassung, dass ein sorgfältiger Fokus auf die Strukturdaten von Fragmenten ein wichtiger Ausgangspunkt für eine erfolgreiche Verknüpfung/Fusion sein könnte, der vorteilhafte Eigenschaften gewährleistet. Wir untersuchen diese Hypothese anhand einfacher Chemie mithilfe eines Virulenzfaktor von Pseudomonas aeruginosa.
P. aeruginosa ist ein Gram-negatives Bakterium, das von der Weltgesundheitsorganisation zu den kritischsten Krankheitserregern gezählt wird.4 Dieses opportunistische Bakterium verursacht ca. 10% der Krankenhausinfektionen und tritt besonders häufig bei Patienten mit geschwächtem Immunsystem und Mukoviszidose auf.5-8 Es hat eine besonders geringe Durchlässigkeit der äußeren Membran, die das Eindringen von Antibiotika in die Zelle verhindert.9 Außerdem transportieren seine Effluxpumpen unerwünschte antimikrobielle Substanzen effizient aus der Zelle heraus, und seine β-Laktamasen sind in der Lage, die entsprechenden β-Laktam-Antibiotika zu inaktivieren,10-13 was zum Ausbruch arzneimittelresistenter P. aeruginosa-Stämme beiträgt.15
Auf der Suche nach Antiinfektiva mit neuartigen Wirkmechanismen ist die Bekämpfung der bakteriellen Virulenz zu einer weit verbreiteten Methode geworden.16-18 Virulenzfaktoren werden von pathogenen Bakterien verwendet und wirken durch verschiedene Mechanismen, darunter das Eindringen in Wirtszellen, die Bildung von Biofilmen und die Umgehung der Immunreaktion des Wirts.19 Die Hemmung von Virulenzfaktoren verringert die bakterielle Virulenz und ermöglicht die Beseitigung der Erreger entweder durch das Immunsystem des Wirts oder durch Antibiotika.20, 21 Die Hauptvorteile dieser Strategie sind der geringere Selektionsdruck auf die Bakterien, wodurch das Risiko einer Resistenzentwicklung sinkt und die Tatsache, dass kommensale Bakterien nicht beeinträchtigt werden.20 Neben anderen Virulenzfaktoren wurde die sekretierte Metalloprotease LasB als eine der wichtigsten Komponenten validiert, die zur Virulenz von P. aeruginosa beitragen.22 LasB ist daher ein besonders attraktives Zielprotein, und wenn man es adressiert, umgeht man Permeations- und Efflux-Probleme aufgrund seiner extrazellulären Lokalisation.
LasB spielt eine entscheidende Rolle bei der pathogenen Invasion von Geweben und ist in erster Linie für akute nosokomiale Infektionen verantwortlich.19, 23 Sie ist in der Lage, Elastin, einen wichtigen Bestandteil von Lungengewebe, Blutgefäßen und Haut, abzubauen, was sie zu einem wichtigen therapeutischen Zielprotein macht. Bislang wurden Naturstoffe wie der Streptomyces-Metalloprotease-Inhibitor (SMPI) aus Streptomyces nigrescens TK-2324 und Phosphoramidon (Pam)25 (Abbildung 1, Verbindung 1), sowie kleine Peptide mit metallchelatbildenden Motiven wie Thiol,26-28 Hydroxamsäure29 oder Carbonsäuren30, 31 beschrieben (Abbildung 1, Verbindung 2). Die meisten dieser Inhibitoren weisen jedoch eine schlechte Selektivität in Bezug auf Säugetier-Metalloenzyme auf. Die kleinen synthetischen Moleküle mit Hydroxamsäure- und Mercaptoacetamid-Gruppen, über die wir berichtet haben (Abbildung 1, Verbindungen 3 und 4), sind vielversprechende LasB-Inhibitoren mit besseren Selektivitätsprofilen, doch sind auf dem Weg zu klinisch anwendbaren Wirkstoffen noch erhebliche Verbesserungen erforderlich.32, 33

Strukturen von bekannten LasB-Inhibitoren. Die zinkbindenden Reste sind hervorgehoben.
Wir haben uns vorgenommen, alternative Bindungsmodi des ursprünglichen Hits zu nutzen, um ein effizientes Zusammenführen und Wachsen der Fragmente zu ermöglichen und die oben erwähnten Einschränkungen der veröffentlichten Inhibitoren zu überwinden. Wir stellen die strukturbasierte Optimierung eines fragmentähnlichen LasB-Inhibitors vor, die zu einer zwölffachen Steigerung der Aktivität in Verbindung mit einer verbesserten in vivo-Wirksamkeit führte. Die inhibitorische Potenz wurde in vitro bestimmt, wobei sichergestellt wurde, dass die Derivate keine direkte antimikrobielle Aktivität aufweisen. Nachdem wir die hervorragende Selektivität anhand einer Reihe repräsentativer menschlicher Off-Targets bestätigt hatten, analysierten wir die Wirksamkeit in vivo in Galleria mellonella-Larven. Eine Kristallstruktur des LasB-Inhibitor-Komplexes bestätigte den vorhergesagten Bindungsmodus.
Wir haben kürzlich die Kristallstruktur der Hit-Verbindung 4 (IC50=6.6±0.3 μM) im Komplex mit LasB veröffentlicht.33 Zu unserer Überraschung befanden sich zwei Moleküle von 4 in der Substratbindungstasche von LasB, was die Voraussetzungen für eine rationale Optimierung der Verbindung durch Zusammenführen beider Moleküle von 4 schuf. Unsere Versuche, diese beiden Moleküle zu einem N-Benzylamid-Derivat zu kombinieren, brachten nicht die gewünschte Aktivitätsverbesserung.33 In unserem nächsten Versuch, die molekularen Wechselwirkungen in der Bindungstasche zu optimieren, verlagerten wir die Benzylgruppe in die Alpha-Position der Amidbindung, um ein nicht-peptidisches Substratimitat (Phe-Seitenkette) zu schaffen, wodurch das erste Derivat der α-Alkyl-N-Aryl-Mercaptoacetamid-Klasse entstand. Verbindung 7 a zeigte eine zweifache Steigerung der Aktivität (Tabelle 1).
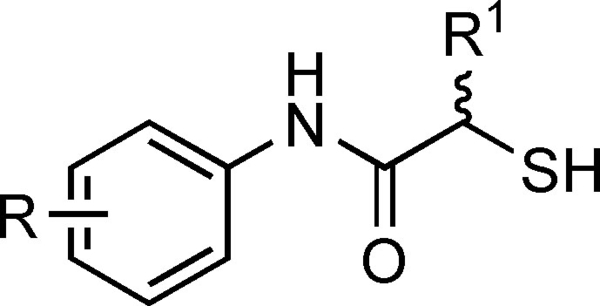
Verbindung |
R |
R1 |
IC50 [μM] |
|---|---|---|---|
4 |
3,4-di-Cl |
H |
6.6±0.3 |
7 a |
3,4-di-Cl |
Benzyl |
2.7±0.4 |
7 b |
3,4-di-Cl |
Cyclohexylmethyl |
12±3 |
7 c |
3,4-di-Cl |
Cyclopropylmethyl |
6.3±1.2 |
7 d |
H |
Benzyl |
1.2±0.1 |
7 e |
2-Me |
Benzyl |
2.4±1.0 |
7 f |
3-Me |
Benzyl |
1.0±0.4 |
7 g |
4-Me |
Benzyl |
0.48±0.04 |
- [a] Mittelwerte±Standardabweichungen von mindestens zwei unabhängigen Experimenten.
Um herauszufinden, inwieweit die Benzylgruppe für den Anstieg der Aktivität verantwortlich ist, haben wir die Wirkung von Cyclohexylmethyl- und Cyclopropylmethyl-Seitenketten weiter untersucht, wobei der di-Chlor-Substituent am N-Arylring erhalten blieb. Obwohl der IC50-Wert von Verbindung 7 c ähnlich wie bei Verbindung 4 mit einer relativ kleinen Cyclopropylmethylgruppe blieb, stieg er bei Verbindung 7 b mit einem Cyclohexylmethylsubstituenten deutlich an (Tabelle 1). Daraus schlossen wir, dass sowohl die optimale Füllung des unbesetzten Raums als auch die Aromatizität der Benzylgruppe zur Aktivität beitrugen.
Unser nächster Schritt bestand darin, die Bedeutung des Di-Chlor-Motivs zu untersuchen, das an wichtigen hydrophoben Wechselwirkungen in der Bindungstasche beteiligt ist. Im Vergleich zu 7 a zeigte Verbindung 7 d eine weitere zweifache Verbesserung des IC50-Werts, was zeigt, dass das Vorhandensein eines di-Chlor-Substituenten nicht wesentlich für die Aktivität der α-Benzyl-N-Aryl-Mercaptoacetamid-Klasse ist.
Um den Bindungsmodus von α-substituierten Mercaptoacetamiden aufzuklären, kristallisierten wir Verbindung 7 d mit LasB (Abbildung 2A). Ausführliche Angaben zur Datenerhebung und zu den Verfeinerungsstatistiken sind in den Hintergrundnformationen (Tabelle S1) zu finden. Wie erwartet, wurde die Verbindung in der gleichen Bindungstasche gefunden, wie sie zuvor für N-(3,4-Dichlorphenyl)mercaptoacetamid-Hit 4 berichtet wurde, und besetzte die S1′–S2′-Taschen mit der Thiolgruppe, die das Zn2+-Kation der aktiven Tasche koordiniert (Abbildung 2B, Abbildung S1). Die N-Arylacetamidgruppe wird durch H-Bindungen und hydrophobe Wechselwirkungen stabilisiert (Abbildung S2). Der Ligand ist möglicherweise in der Bindungstasche durch den Carbonylsauerstoff verankert, der eine bidendate Wasserstoffbrückenbindung mit Arg198 bildet. Die Phenylgruppe des N-Arylacetamids besetzt den weiten, offenen und für Lösungsmittel zugänglichen Eingang der S1′-Bindungstasche, was die beiden unterschiedlichen Ausrichtungen erklären könnte, die für diesen Teil der Verbindung in der Kristallstruktur beobachtet wurden (Abbildung 2). Die Benzylgruppe liegt in der lipophilen S2′-Bindungstasche und wird durch zahlreiche hydrophobe Wechselwirkungen stabilisiert (Abbildung S2).

A Kristallstruktur von LasB im Komplex mit 7 d (PDB-Code: 7OC7). Cartoon-Darstellung von LasB (hellblau) im Komplex mit 7 d (schwarz). Das graue Isomesh stellt die Polderkarte von 7 d dar, die bei 2 σ konturiert ist. Es werden zwei verschiedene Zustände von 7 d mit unterschiedlichen Besetzungen beobachtet. B Überlagerung der Strukturen von LasB (hellblaue Oberfläche) im Komplex mit 4 (gelb) oder 7 d (schwarz, Hauptkonformation dargestellt), die die Phenylgruppe zeigt, die die S1′-Bindungsstelle des Enzyms besetzt. Das Zn2+-Kation im aktiven Zentrum ist als graue Kugel dargestellt.
Im Gegensatz zum Bindungsmodus von Verbindung 4 bindet nur ein Molekül von Verbindung 7 d an das Protein, was zur Schließung der Bindungstasche führt - ein Phänomen, das allgemein bei Thermolysin-ähnlichen Proteasen wie LasB nach der Bindung eines Inhibitors beobachtet wird.34 Unsere strukturbasierte Strategie, den Benzylrest vom Amidstickstoff in die Alpha-Position zu verlagern, erwies sich als erfolgreich, da wir den Platz in der Bindungstasche mit einem Molekül besetzen konnten.33
Die Kristallstruktur von LasB–7 d ermöglichte ein tieferes Verständnis der potenziellen Wechselwirkungen im umgebenden unbesetzten Raum und ebnete den Weg für weitere Optimierungen. So ist beispielsweise die Toleranz gegenüber anderen lipophilen Substituenten, insbesondere in der S1′-Tasche, gut dokumentiert. Da wir zuvor festgestellt hatten, dass das hydrophobe Di-Chlor-Motiv für die Verbesserung der Aktivität nicht wesentlich ist, war unsere nächste Wahl für einen lipophilen Substituenten eine sterisch weniger anspruchsvolle Methylgruppe. Wir haben die Kristallstruktur für eine gezielte, strukturbasierte Optimierungsstudie verwendet. Wie aus der Dockingpose (Abbildung S3) hervorgeht, führt das Vorhandensein eines lipophilen Methylsubstituenten in der para-Position höchstwahrscheinlich zu einer weiteren Verstärkung der hydrophoben Wechselwirkungen mit Leu197 und bietet in Kombination mit der Benzylgruppe in der alpha-Position optimale Wechselwirkungen in der Bindungstasche. Vor diesem Hintergrund haben wir drei Regioisomere synthetisiert, die sich alle als wirksamer erwiesen als die ursprünglich optimierte Struktur, Verbindung 7 a (Tabelle 1). Erwartungsgemäß hatte die Einführung eines para-Methyl-Substituenten am aromatischen Kern der N-Arylacetamid-Gruppe die stärkste Auswirkung auf die Aktivität, was die günstigen Inhibitor–Protein-Wechselwirkungen bestätigt. Diese Wechselwirkungen sind der Grund für die submikromolare Aktivität von Verbindung 7 g (IC50=0.48±0.04 μM).
Der Syntheseweg für alle Derivate ist in Schema 1 dargestellt. Durch Diazotierung und anschließende Chlorierung der entsprechenden kommerziell erhältlichen racemischen Aminosäuren 4 a–4 c wurden α-Chlorcarbonsäuren erhalten. Deren Kopplung mit dem jeweiligen Anilin die Zwischenprodukte 5 a–5 g ergab. Die Thioacetatfunktion wurde durch eine SN2-Reaktion mit Kaliumthioacetat 6 a–6 g eingeführt. Die Entschützung des Thioacetats unter basischen Bedingungen ergab die Endverbindungen 7 a–7 g in 20–88% Ausbeute als freie Thiole.

Syntheseschema der Klasse der α-Alkyl/Aryl-Verbindungen.[a]
Bevor wir die in vivo Wirkung unserer verbesserten Inhibitoren untersuchten, analysierten wir ihre antibakterielle Aktivität. Ein Test der minimalen Hemmkonzentration (MHK) ergab keine unmittelbare Auswirkung des Inhibitors 7 d auf das Wachstum des Pathogens (Hintergrundinformationen, Seite S3, MHK>100 μM). Darüber hinaus untersuchten wir seine Zytotoxizität gegenüber drei menschlichen Zelllinien (Hintergrundinformationen, Seite S3, IC50>100 μM). Da eine geringe Selektivität gegenüber Metalloproteasen häufig ein Problem darstellt, untersuchten wir die Selektivität ausgewählter LasB-Inhibitoren gegenüber sechs Matrix-Metalloproteasen (MMPs) und drei weiteren humanen Off-Targets (Tabelle S2). Während die Selektivität der optimierten Verbindungen 7 d und 7 g im Vergleich zur Hit-Verbindung 4 mit Ausnahme von TACE beibehalten werden konnte, wurde eine leicht verringerte Zytotoxizität der neuen Verbindungen nachgewiesen.
Anschließend analysierten wir die Antivirulenz-Aktivität von LasB-Inhibitoren in vivo anhand eines einfachen Modells, das auf G. mellonella-Larven basiert. Diese Methode dient dazu, die Behandlungsmöglichkeiten für P. aeruginosa-induzierte Infektionen zu bewerten und die Wirksamkeit von LasB-Inhibitoren in Vorbereitung auf eine in vivo-Pharmakodynamikstudie an Mäusen unter Beweis zu stellen.32, 33 Wir injizierten den Larven den Überstand (s.n.) von P. aeruginosa PA14 mit oder ohne 0.25 nmol der Verbindungen 4 oder 7 g, inkubierten sie drei Tage lang und erfassten die Überlebensrate einmal pro Tag (Abbildung 3). Unsere Ergebnisse zeigten, dass PA14 s.n. die Überlebensrate der Larven nach drei Tagen Inkubation um bis zu 88% reduzierte. Verbindung 4, die als Kontrolle verwendet wurde, zeigte nach drei Tagen praktisch keine Verbesserung der Überlebensrate. Die Behandlung der Larven mit 0.25 nmol der Verbindung 7 g erhöhte die Überlebensrate um bis zu 60% im Vergleich zu den mit PBS injizierten Larven. Diese Ergebnisse validieren unsere Inhibitoren als vielversprechende Kandidaten zur Blockierung der Pathogenität von P. aeruginosa und bestätigen, dass die Steigerung der hemmenden Aktivität in vitro auch zu einer verbesserten Wirkung in vivo führt.

Kaplan-Meier-Überlebensanalyse von G. mellonella Larven, die mit 0.25 nmol Verbindung 4 (getüpfelt) (p≤0.9452), 0.25 nmol Verbindung 7 g (gestrichelt) (p≤0.0002) oder nur PA14 s.n. (durchgehend) behandelt wurden (n=3). PA14 s.n. war auch während der Behandlung mit der Verbindung enthalten. PBS diente als Negativkontrolle (getüpfelt und gestrichelt). Die Verbindungen 7 g und 4 in PBS zeigten eine 100%ige Überlebensrate. s.n.: Überstand.
Zusammenfassend lässt sich sagen, dass wir durch strukturgeleitete Fragment-Fusion/Linking, das sich am natürlichen Substrat inspiriert, eine erhebliche Steigerung der Potenz unserer LasB-Inhibitoren erreicht haben, die sich in einer in vivo-Wirksamkeit niederschlägt. Wir haben die Verbindung 7 g identifiziert, die eine zwölffache Verbesserung der Aktivität im Vergleich zu unserem besten zuvor beschriebenen Inhibitor aufweist. Ermutigt durch die ausgezeichnete in vitro-Aktivität dieser Verbindungen haben wir auch eine in vivo-Wirkung in einem G. mellonella-Modell nachgewiesen. Die Überlebensrate von Larven, die mit PA14-Überstand infiziert und mit Verbindung 7 g behandelt wurden, war im Vergleich zu unserem vorherigen Hemmstoff 4 deutlich verbessert.
Obwohl die strukturgeleitete Erforschung der N-Arylringsubstitution uns einen guten Ausgangspunkt für die Leitstrukturoptimierung lieferte, sollte eine systematische Variation des Substituenten in der α-Position (R1) in Betracht gezogen werden, um die Lipophilie der S2′-Tasche zu nutzen. Darüber hinaus nimmt die Verbindung 7 g nur einen kleinen Teil (ca. 24%) des gesamten vorhergesagten Volumens der Bindungstasche ein und lässt die S2′-Tasche weitgehend unberührt (Abbildung S4). Daher sollte eine Erweiterung der Thiolgruppe in diese Richtung zusätzliche Ligand–Protein-Wechselwirkungen ermöglichen, was zu einer weiteren Verbesserung der hemmenden Wirkung führen sollte. Die Berechnung der Ligandeneffizienz (LE) und der lipophilen Ligandeneffizienz (LLE) für Verbindung 4 ((LE:0.44, LLE:1.67) und unserem optimierten Hit 7 g (LE:0.43, LLE:2.37) ergab eine leichte Verbesserung der LLE bei gleichbleibender LE, was einen erfolgreichen Ausgangspunkt für die weitere Optimierung und Feinabstimmung dieser Klasse darstellt.
Unsere Arbeit zeigt, wie wichtig es ist, alternative Bindungsmodi zu nutzen, um eine einfache Fragmentverschmelzung zu erreichen. Das vom Substrat inspirierte Design führte zu einer verbesserten Potenz der zuvor identifizierten Strukturen bei gleichzeitiger Steigerung der Wirksamkeit in vivo und beschleunigte so den Weg der translationalen Entwicklung. Dieses Konzept sollte auch auf andere Zielptoteine übertragbar sein und stellt eine wichtige Grundlage für die künftige Entwicklung dieser Klasse von Inhibitoren dar, da sie das Potenzial haben, vielversprechende Kandidaten für den therapeutischen Einsatz zu werden und den Konzeptnachweis für kleine synthetische Moleküle zu erbringen, die auf Virulenzfaktoren in der Klinik abzielen.
Danksagung
Die Autoren sind für die technische Unterstützung durch Simone Amann, Jeannine Jung, Selina Wolter und Dennis Jener dankbar. A.K.H. Hirsch bedankt sich für die Förderung durch den Impuls- und Vernetzungsfonds der Helmholtz-Gemeinschaft. J. Konstantinović bedankt sich für die Förderung durch die Alexander von Humboldt-Stiftung. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.

