

Characterization of Mannosyl Dioxanium Ions in Solution Using Chemical Exchange Saturation Transfer NMR Spectroscopy
Abstract
The stereoselective introduction of the glycosidic bond remains one of the main challenges in carbohydrate synthesis. Characterizing the reactive intermediates of this reaction is key to develop stereoselective glycosylation reactions. Herein we report the characterization of low-populated, rapidly equilibrating mannosyl dioxanium ions that arise from participation of a C-3 acyl group using chemical exchange saturation transfer (CEST) NMR spectroscopy. Dioxanium ion structure and equilibration kinetics were measured under relevant glycosylation conditions and highly α-selective couplings were observed suggesting glycosylation took place via this elusive intermediate.
The stereoselective introduction of glycosidic bonds (glycosylation) is one of the main challenges in the chemical synthesis of carbohydrates.1 In a chemical glycosylation reaction, an electrophile (the glycosyl donor) is activated by a chemical promotor and reacts with a nucleophile (the glycosyl acceptor). The nucleophile can add from the α- or β-face to reactive intermediates thereby leading to the formation of α- or β-diastereoisomers, respectively. The stereochemical outcome of glycosylation reactions can be controlled using neighbouring group participation (NGP) of C-2 acyl groups of glycosyl donors.2 Upon activation of such glycosyl donors, the C-2 acyl group can engage in NGP affording bicyclic dioxolenium ion intermediates that react in a stereospecific manner with glycosyl acceptors to afford 1,2-trans products.3 Extension of this principle to acyl functionalities positioned on the C-3, C-4 or C-6 hydroxyl groups via NGP has also been suggested to direct the stereoselectivity of glycosylation reactions. However, whether selectivity can be attributed to NGP of the ester group or other stereoelectronic effects is a subject of much debate.4 Due to the instability of the intermediate glycosyl cations that result from participation of C-3, C-4 or C-6 acyl groups, it is exceedingly difficult to detect and characterize them and has only been achieved for the more stable dioxolenium ions resulting from NGP of a C-2 acyl group.4a, 5 Indirect evidence for NGP of more remote positions than C-2 has been provided by trapping intermediates resulting from acetyl ester, carbonate or imidate NGP to provide a bicyclic orthoester,6 carbonate7 or oxazolines,8 respectively.4c Recently, we reported the first spectroscopic evidence of bridged intermediates resulting from the NGP of C-3 and C-4 esters in the gas phase using infrared ion spectroscopy (IRIS).4d We found that the participation of acyl groups positioned at C-3 of mannose was most favorable. Solution-phase experiments showed that glycosylation of mannosides carrying a single acyl group at C-3 proceeded with very high α-selectivity compared to derivatives containing a non-participation group at C-3. This observation is consistent with NGP of the C-3 ester thereby shielding the β-face and only allowing nucleophilic addition to the α-face. Although DFT calculations modeling solvent conditions showed that C-3 NGP was favorable, direct experimental evidence demonstrating that C-3 NGP occurs in solution is still lacking. The absence of a counter ion in the gas phase allows for the characterization of “naked” glycosyl cations but is much more challenging to achieve in solution due the rapid equilibrium of the dioxanium ion 2 with the more stable α-glycosyl triflate 3 (Scheme 1 A). For molecules such as mannoside 1 we do not expect α-glycosyl triflates to be the reactive intermediates directly leading to observed α-glycoside products as they are known to react with inversion of stereochemistry via an SN2-like mechanism.4c Instead, an α-selective reaction proceeding via a higher energy product intermediate such as dioxanium ion 2 resulting for the NGP of the C-3 ester is expected according to the Curtin–Hammett principle.

A) Structure and expected stereoselectivity of the equilibrating dioxanium ion and α-glycosyl triflate. Detection of the minor dioxanium ion population via saturation tranfer to the major α-glycosyl triflate species via chemical exchange. B) Structure of the 3-O-acyl protected glycosyl donor used in the study.
Inspired by recent work reported by Gschwind and co-workers on the characterization of iminium ions,9 we reasoned that the detection of low abundance dioxanium intermediates could be possible via chemical exchange saturation transfer (CEST) NMR. This technique has been widely applied in the (bio)chemical NMR community using high magnetic fields to detect high-energy or “invisible” states that are in chemical exchange with a highly-populated, visible ground state.10 No prior information about the chemical shift of the low-populated intermediate resonance is required since the experiment scans a given frequency domain by incrementing the saturation offset frequency whilst monitoring the transfer of saturation to the main observable species via chemical exchange.
Herein we report the use of CEST-NMR to provide the first spectroscopic evidence for NGP of C-3 esters on mannosides under relevant glycosylation conditions. Furthermore, by varying saturation times, the exchange kinetics were measured and using simulations, we reasoned that reactions on the dioxanium ion and corresponding α-triflate fall within the boundaries expected for a Curtin–Hammett scenario, which plausibly explains the α-selective nature of these mannosyl donors.
The detection of chemical exchange via saturation transfer is possible if the chemical exchange is slow on the NMR timescale (k1+k−1≤Δω). Furthermore, the detection window is defined by a combination of relative population exchanging species, longitudinal relaxation rate (R1), and exchange rates (k1 and k−1).11 Hence, to study NGP of the mannosyl 3-O-acyl group, we designed and prepared 13C-labeled donors 1, 4–6 (Scheme 1 B) to increase the sensitivity of the 13C CEST experiments. 13C-enriched probes were installed on the 3-O-acyl group as the anticipated dioxanium quaternary carbon would be shifted significantly in frequency (larger Δω) due to build-up of positive charge. Labelling the carbonyl also had the added benefit that carbonyl 13Cs typically have much smaller R1s compared to their CH counterparts due to the absence of attached 1Hs to assist in dipolar relaxation. Methyl ether protecting groups were used to reduce R1 due to molecular tumbling compared to molecules carrying benzyl protecting groups. Based on IRIS, DFT and solution-based experiments of glucosyl donors such as 6, we expect the contribution of NGP to be negligible as these donors display poor stereoselectivity with a variety of nucleophiles. Hence, we also prepared glucosyl donor 6 as a negative control. CEST-NMR experiments with 1 and 4–6 were performed using activation with diphenyl sulfoxide (Ph2SO) and triflic anhydride (Tf2O) in DCM-d2 at −80 °C,12 heated to −40 °C until the initially formed α-oxosulfonium ion was consumed (1–1.5 h) and subsequently cooled back to −80 °C. Experiments were performed either in presence or the absence of the non-nucleophilic base 2,4,6-tri-tert-butylpyrimidine (TTBP).
Activation of donor 1 under these conditions in the absence of TTBP led to the formation of three species that were detected in the 13C 1D NMR spectrum (Figure S1). From these species, α-glycosyl triflate 3 could be identified. No evidence for the formation of the 1,3-bridged dioxanium ion (2) was visible in the 13C 1D spectrum. Next, CEST-NMR was used to scan the region from δC=200 to 160 ppm by sweeping with a saturation field of 40 Hz and detecting sparsely-populated exchanging intermediates via saturation transfer to the 13C peak corresponding to the α-glycosyl triflate 3 (δC=171 ppm) as a function of frequency. To our delight, even though its population is too low to allow for direct detection in the 13C 1D spectrum, the CEST-spectrum revealed the presence of a species at δC=187 ppm that is exchanging with 3 (Figure 1). Based on the typical 13C-chemical shift of a dioxolenium ion,13 this species was reasoned to be a dioxanium ion 2. Notably, the other minor species present in the 13C 1D spectrum did not show exchange with the α-glycosyl triflate 3. Surprisingly, a heating (−40 °C), stabilizing (2 h), and cooling (−80 °C) cycle clearly showed a detectable population of the dioxanium ion (Figure 1 B). Unfortunately, efforts to characterize the dioxanium ion based on HMBC-NMR failed due to its rapid exchange rate.

A) Activation Scheme for glycosyl donor 1 and the corresponding intermediates expected. B) 13C 1D NMR spectrum (black) overlaid with the CEST-spectrum (red) showing the presence of an low populated intermediate that is exchanging with the α-triflate resonance at δC=171 ppm.
We reasoned that substituting the acetyl ester for a benzoyl ester might increase the corresponding dioxanium ion stability via resonance thereby reducing the fast exchange that limited the HMBC experiment and increase its population. Hence, 4 was activated in the absence of TTBP and a mixture of species was observed (Figures S2–S4) including a clear peak at δC=177.2 ppm corresponding to dioxanium ion 7, and further analysis was performed by HMBC and COSY experiments (Figure 2). The existence of the 1,3-bridged dioxanium ion 7 was confirmed by the presence of a cross-peak from the anomeric proton at δH=6.48 ppm and the labelled carbonyl carbon at δC=177.2 ppm in the HMBC-spectrum (Figure 2). In addition, COSY-NMR showed the three W-patterns present in the dioxanium ion on the basis of coupling between the resonances at δH=6.48, 5.59 and 4.27 ppm. Further characterization of the dioxanium ion was limited by the typically small vicinal axial-equatorial or equatorial-equatorial coupling and peak broadening due to temperature and exchange (Figure S5).

Characterization of the dioxanium ion: HMBC-NMR displays the cross-peak from the anomeric proton at δH=6.48 ppm and the labelled carbonyl carbon at δC=177.2 ppm and COSY-NMR displays the characteristic W-coupling patterns expected for the dioxanium ion in the 1C4-chair.
These observations indicate that the dioxanium ion adopts a 1C4-chair conformation (Figure 2), which is in agreement with the structure found by IRIS.4d CEST-NMR revealed solely exchange of the resonance at δC=177.2 ppm with the α-glycosyl triflate (8) carbonyl peak (Figure S6). Again, heating the sample for 2 h to −40 °C showed an increase in dioxanium ion population (Figure S7). To obtain additional experimental evidence for the presence of 1,3-bridged dioxanium ion 7 we prepared mannosyl donor 5 which contains a 13C label at both the benzoyl group and the anomeric centre. Upon activation of 5 at −40 °C for a few hours and cooling to −80 °C in the absence of TTBP, a vicinal 3JC-C coupling of 2–3 Hz was observed for the labelled dioxanium ion carbons indicating covalent interaction of the 3-O-benzoyl group with C-1 (Figure S8). Finally, by taking advantage of the anomeric 13C-label, we scanned frequencies from δC=280 to 200 ppm by CEST-NMR to probe the presence of the oxocarbenium ion.14 Unfortunately, no evidence for the oxocarbenium ion was found, likely due to the short lifetime that limits detection (Figure S9).15
Having established the formation of the 1,3-bridged dioxanium ion as a result of NGP of the C-3 acyl group with CEST-NMR under conditions lacking TTBP, we studied NGP of the C-3 acyl group in the presence of TTBP, which represents more relevant glycosylation conditions. Interestingly, activation of donor 5 in presence of TTBP showed a more rapid consumption of the α-oxosulfonium ion. The milder conditions showed less side-product formation and a lower population of dioxanium ion 9 was observed after activation. The dioxanium ion could only be directly observed by acquiring 128–512 scans, in addition to signals corresponding to TTBP and various degradation products (Figure 3 B and S10,11). However, the CEST-spectrum clearly showed the presence of the dioxanium ion with just 8 scans (Figure 3 B), highlighting the sensitivity of CEST-NMR.

A) Activation Scheme for glycosyl donor 5 and the corresponding intermediates. B) 13C 1D NMR spectrum (black) overlaid with the CEST-spectrum (red) showing the presence of an invisible intermediate that is exchanging with the α-triflate resonance at δC=165 ppm. C,D) Saturation decay plots displaying the effect of varying saturation times at the dioxanium ion to the intensity of the α-triflate resonance.
To obtain extra information about the exchange kinetics of the α-triflate (10) and dioxanium ion (9), CEST-NMR was used to derive the exchange rate constant from the α-triflate to the dioxanium ion (kαd). This was derived from the 13C-intenstity of the α-triflate resonance upon varying the saturation time at the dioxanium ion frequency (see SI for experimental details). From the data presented in the saturation decay curves (Figures 3 C and D) was calculated kαd of 0.98 s−1 and 0.95 s−1 in the situations without and with TTBP, respectively. As Tf2O was used without further purification, a small, unknown amount of water and triflic acid (TfOH) is present. This unfortunately makes it difficult to estimate the equilibrium constant (Keq) and the exchange rate constant from the dioxanium ion to the α-triflate (kdα) in experiments without TTBP (Figure S12B,C). However, in the presence of TTBP, free triflate ([−OTf]) can be more accurately accounted for because the excess base will react with any TfOH or hydronium triflate, and TTBPH+ also has unique 1H NMR signals (Figure S12D–F). Based on these experiments Keq and kdα were determined under the glycosylation conditions to be 437 M−1 and 415 s−1 M−1, respectively (Figure 3 A).
Recently, gas-phase experiments have confirmed NGP also in 3-O-acyl glucosides and a slight increase in stereoselectivity was observed. However, this effect was shown to be much less pronounced than in 3-O-acyl mannosides which led us to investigate if there is also a difference in glycosylation intermediates observed by CEST-NMR. Hence, glucosyl donor 6 was investigated for NGP and activated both with and without TTBP. In contrast to mannosyl donor 4, NMR experiments revealed no evidence for the formation of a dioxanium ion, even after a heating cycle (Figure S13).
To ensure that both experiments with mannosyl donor 4 and glucosyl donor 6 were conducted under representative glycosylation conditions, activated glycosyl donors 4 and 6 were treated with ethanol (EtOH) or 2,2,2-trifluoroethanol (TFE). All reactions were finished within five minutes after addition of the nucleophile at −80 °C (Figures S14–S17). Mannosyl donor 4 was selective towards the α-product for both acceptors, whereas glucosyl donor 6 was much less stereoselective (Table 1). The observed selectivity matched well with earlier findings.4d It is possible that other reactive intermediates are present in both cases but cannot be detected using CEST-NMR as the kinetics of the chemical equilibrium needs to fall within the aforementioned boundary conditions for CEST-NMR to enable their detection. However, the presence of dioxanium ions in the manno case and absence in the gluco case reflects the difference in stereoselectivity and suggests a role for dioxanium ion as a reactive intermediate. Finally, we compared tetra-O-methyl mannosyl donor 11 to 3-O-acyl-mannosyl donor 4. In previous studies, 11 was shown to give varying selectivity depending on the nucleophilic strength of the acceptor (Table 1).4d Therefore, the comparative data also suggest a role for the dioxanium ion as a reactive intermediate as 4 consistently delivers high α-selective coupling irrespective of the nucleophilic strength of the acceptor.
Entry |
Donor |
Acceptor |
Selectivity[a] (α/β) |
|---|---|---|---|
1 |
|
EtOH |
51/49[b] |
2 |
TFE |
93/7[b] |
|
3 |
|
EtOH |
100/0 |
4 |
TFE |
100/0 |
|
5 |
|
EtOH |
25/75 |
6 |
TFE |
73/27 |
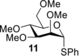
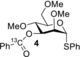
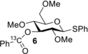
- Conditions: i) glycosyl donor 4 or 6, Ph2SO (1.1 equiv.), Tf2O (1.3 equiv.), TTBP (2.5 equiv.), −80 °C, CD2Cl2; ii) −40 °C, 1–1.5 h; iii) −80 °C, either EtOH or TFE (2.0 equiv.) [a] On the basis of 1H- and HSQC-NMR (Figures S18–S21). [b] Literature values; Conditions: i) glycosyl donor 11, Ph2SO (1.3 equiv.), Tf2O (1.3 equiv.), TTBP (2.5 equiv.), −80 °C, CH2Cl2; ii) −40 °C, 1–1.5 h; iii) −80 °C, either EtOH or TFE (2.0 equiv.).4d
A plausible explanation for the observed selectivity is that the reaction is operating under the Curtin–Hammett principle whereby the high-energy dioxanium ion determines selectivity with the interconversion between intermediates 7 and 8 occurring much faster than the reaction of the α-triflate with the nucleophile. Given the rate constants, kαd and kdα, above and literature rate constants for nucleophilic attack of TFE on glycosyl α-triflates, reaction time-course simulations were performed to test this hypothesis.16 Simulations employing conservative estimates for the rate constants describing nucleophilic attack on 7 and 8 clearly indicate that interconversion of the α-triflate to dioxanium occurs significantly faster than nucleophilic attack on the α-triflate, supporting Curtin–Hammett when using donor 4 (Figure S22–S25).
In conclusion, we present direct experimental evidence for the NGP of 3-O-acyl groups on mannosides using CEST-NMR. The kinetics of the chemical exchange between the α-glycosyl triflate and dioxanium was established under relevant glycosylation conditions. NGP of a 3-O-acyl groups was not detected in the corresponding glucose derivative consistent with its lower α-selectivity. These findings highlight the potential importance of low populated reaction intermediates. Additionally, CEST-NMR was established as useful tool to study the chemical glycosylation reaction and shows that the technique can conveniently be used with low field magnets, making it a very accessible tool.
Acknowledgements
This work was supported by a VIDI grant (192.070) and ERC-Stg (Glycoedit, 758913) awarded to T.J.B.
Conflict of interest
The authors declare no conflict of interest.

