Additional three patients with Smith-McCort dysplasia due to novel RAB33B mutations
Abstract
Smith-McCort dysplasia (SMC OMIM 615222) and Dyggve-Melchior-Clausen dysplasia (DMC OMIM 223800) are allelic skeletal dysplasias caused by homozygous or compound heterozygous mutations in DYM (OMIM 607461). Both disorders share the same skeletal phenotypes characterized by spondylo-epi-metaphyseal dysplasia with distinctive lacy ilia. The difference rests on the presence or absence of intellectual disability, that is, intellectual disability in DMC and normal cognition in SMC. However, genetic heterogeneity was suspected in SMC. Recently, RAB33B (OMIM 605950) has been identified as the second gene for SMC. Nevertheless, only two affected families have been reported so far. Here we present three SMC patients with four novel pathogenic variants in RAB33B, including homozygosity for c.211C>T (p.R71*), homozygosity for c.365T>C (p.F122S), and compound heterozygosity for c.48delCGGGGCAG (p.G17Vfs*58) and c.490C>T (p.Q164*). We also summarize the clinical, radiological, and mutation profile of RAB33B after literature mining. This report ascertains the pathogenic relationship between RAB33B and SMC. © 2017 Wiley Periodicals, Inc.
INTRODUCTION
Smith-McCort dysplasia (SMC) and Dyggve-Melchior-Clausen syndrome (DMC) are rare autosomal recessive skeletal dysplasias, which share common skeletal phenotypes, characterized by spondylo-epiphyseal-metaphyseal dysplasias (SEMD), lacy ilia, and other distinctive skeletal changes. The two conditions are only distinguishable by presence of intellectual disability in DMC but normal cognition in SMC [Spranger et al., 1976]. Analyses for patients with SMC and DMC revealed linkage to loci on chromosome 18q12 and then mutations in DYM gene in both disorders [Ehtesham et al., 2002; El Ghouzzi et al., 2003; Ahmad et al., 2010]. Till date, 46 patients from 30 families with DMC and 8 patients from 3 families with SMC were reported with a mutation in DYM [Burns et al., 2003; Cohn et al., 2003; El Ghouzzi et al., 2003; Paupe et al., 2004; Kinning et al., 2005; Pogue et al., 2005; Neumann et al., 2006; Santos et al., 2009; Denais et al., 2011; Khalifa et al., 2011]. However, mutations in another gene, RAB33B, are also known to cause SMC and until now only five patients from two families have been reported with mutations in this gene [Alshammari et al., 2012; Dupuis et al., 2013]. RAB33B protein belongs to small GTPase superfamily which is known to be involved in Golgi trafficking and autophagy [Zheng et al., 1998; Itoh et al., 2008; Starr et al., 2010]. Here, we describe three patients with SMC due to mutations in RAB33B gene which fortifies the previous findings, and also confirm its clinical features.
MATERIALS AND METHODS
Appropriate informed consents were obtained from the families for this study. Detailed clinical presentations, radiological findings and family history were recorded. Genomic DNA was isolated from blood collected from three families using QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA) according to manufacturer's instructions. All coding exons and flanking introns of DYM gene (NM_017653.3) and RAB33B gene (NM_031296.1) were amplified by PCR using specifically designed primers. PCR amplicons were purified and sequenced by ABI PRISM 3130 genetic analyzer (Life Technologies, Foster City, CA). Multiple sequence alignment and in silico prediction of pathogenicity were analyzed using bioinformatics tools MutationTaster, SIFT, and PolyPhen2. The study protocol has the approval of the respective institutional ethics committee.
RESULTS
We recruited two patients from India (patients 1 and 2) and one patient from South Korea (patient 3) along with their parents for this study.
Clinical Reports
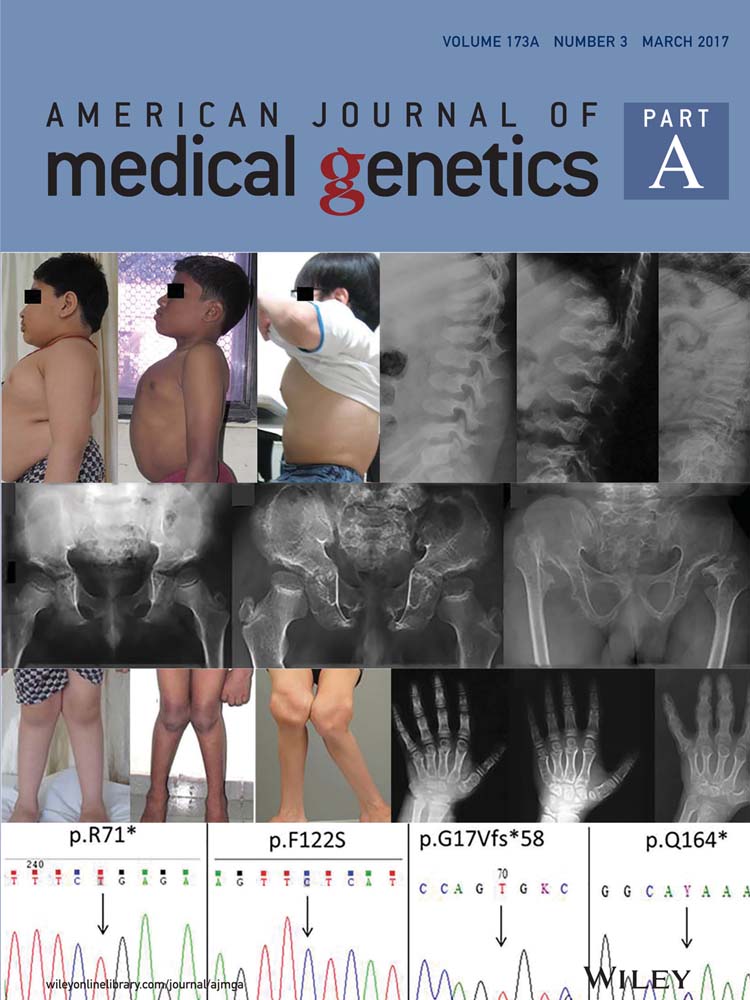
Patient 1, 7-year-old male child, presented with short stature, chest deformity, recurrent episodes of lower respiratory tract infections and frequent episodes of vomiting. He had an abnormal gait and was not able to squat. Progressive difficulty in walking was also noticed since the age of 3 years. He was the third child to a non-consanguineous couple with no other significant family history. He was born at full term by normal vaginal delivery and had a birth weight of 2.5 kg (2 SD below the mean). His developmental milestones were normal. On examination at age 7 years, his height was 110.5 cm (2 SD below the mean), head circumference was 52 cm (normal), weight was 32 kg (2 SD above the mean) and upper segment to lower segment ratio was 0.97. His phenotype included a short neck, pectus carinatum, limited extension of elbows and genu valga (Figs. 1A,D and 2D). Brachydactyly and pes planus were also noticed (Fig. 2A and G). There was no organomegaly. Hematology and biochemical investigations were unremarkable.


Patient 2, was the first born to a fourth degree consanguineous couple. Antenatal period was unremarkable. Sonographic antenatal screening at 34–35 weeks revealed no skeletal abnormality. He was born at term by forceps delivery with a birth weight of 2.75 kg. There were no neonatal complications. All motor and sensory milestones were appropriate for his age when assessed at age 4 years. He was investigated for poor weight gain, abnormal walking, and short stature. Initially, he was suspected to have mucopolysaccharidosis IV, which was investigated and was not confirmed. At age 12 years and 9 months, his height was 126 cm (4 SD below the mean), weight was 31.6 kg (2 SD below the mean), and head circumference was 52 cm (normal). He presented small forehead, posteriorly rotated ears, mild prognathism, and facial appearance was similar to Morquio syndrome. He showed severe pectus carinatum, lumbar lordosis, genu valgum, brachydactyly in hands, and overlapping of toes T2 and T3 bilaterally (Figs. 1B,E and 2B, E, and H). Rest of the systemic examination including neuromuscular system, blood counts, serum chemistry were unremarkable.
Patient 3 presented with short stature and marked genu valgum deformity at age 31 years. His birth weight and height were recalled to be within normal limit, but delayed height gain was noticed since 2 years of age. Developmental milestones were within normal limits. He was short but physically active with mild genu valgum until 6 years of age. Then, genu valgum gradually aggravated and he underwent surgical correction by distal femoral osteotomy at age 9 years. He did not have any medical check-up thereafter. Genu valgum recurred and aggravated, and hip joint pain and weakness developed around age 14 years, making walking upstairs difficult and Taylor position impossible (Fig. 2F). At 16 years of age, the patient presented with painful limitation of hip abduction, and was wheelchair-bound. He was 107.5 cm (7–8 SD below mean) and 17.5 kg (5 SD below mean) at age 14 years, and the final height at age 31 years was 115 cm (8–9 SD below mean).
He had myopia, but did not show signs of hearing loss. Physical examination showed increased knee jerk and borderline Babinski signs. Plain radiograph and MRI showed atlantoaxial instability and compressive myelopathy. He could barely stand up with due to externally rotated and flexed hips and the patellae facing laterally. The femorotibial angle was over 50 degrees, but was difficult to determine because of ligamentous laxity. He had thoracolumbar kyphosis and increased anteroposterior distance of the thoracic cage (Fig. 1C,F). Feet and hands were small for the body size (Fig. 2C,I). There was a remarkable resemblance of the clinical and radiographic features to Morquio syndrome. Survey for urinary, enzymatic, and molecular study of MPS IV revealed no abnormalities.
Skeletal survey of all three patients showed predominant abnormalities of the spine and pelvic bones, mildly affected epi-metaphyses of the long bones, and brachydactyly. Presence of platyspondyly exhibiting a central constriction of the upper and lower end-plates of the vertebral bodies, also called “double humped appearance” was seen at an early age but became less distinct with age (Fig. 3A–C).

The iliac bones were small, with tapering lower ends, and thick ischial and pubic bones. A distinctive and diagnostic lacy appearance of the iliac crest was evident but disappeared in adult patient (Fig. 3D–F). Brachydactyly with consistently short metacarpals and dysplastic carpal bones was observed (Fig. 4).

Clinical and radiological features of the current and reported patients with mutations in RAB33B are listed in Table I.
| Present study | Alshammari et al. (2012) | Dupuis et al. (2013); Neumann et al. (2006) | ||||||
|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
| General features | ||||||||
| Age at current evaluation (in years) | 7 | 12 | 36 | NA | 11 | NA | NA | 22 |
| Gender | Male | Male | Male | Female | Male | Female | Female | Male |
| Country of origin | India | India | Korea | Saudi Arabia | Saudi Arabia | Saudi Arabia | Saudi Arabia | Turkey |
| Affected siblings | None | None | None | 3/8 | 3/8 | 3/8 | 3/8 | None |
| Consanguinity | − | + | − | + | + | + | + | + |
| Height (cm)/SD | 110.5/−2 | 126/−3.8 | 115/−9 | 119/−4.3 | 111.3/−4.7 | 121/−2.3 | 90.5/−2.4 | 133/−7.3 |
| Weight (kg)/SD | 32/−2 | 31.6/−2 | 28/−5.3 | 26/−2.5 | 21/−3 | 25.1/ −2 | 13.4/−2 | − |
| Clinical features | ||||||||
| Cognition | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Coarse face | − | + | + | − | − | − | − | + |
| Short neck | + | + | + | + | + | + | + | + |
| Short trunk | + | + | + | NA | + | NA | NA | + |
| Barrel chest and pectus carinatum | + | + | + | + | + | + | + | + |
| Limited extension of elbow joints | + | + | + | + | + | + | + | + |
| Short and broad fingers | + | + | + | + | + | + | + | + |
| Accentuated lumbar lordosis | + | + | − | + | + | + | + | + |
| Genu valgum | + | + | + | + | + | + | + | + |
| Short and broad toes/toe abnormality | + | + | + | NA | NA | NA | NA | NA |
| Waddling gait | + | + | NA | NA | NA | NA | NA | + |
| Radiological features | ||||||||
| Platyspondyly | + | + | + | + | + | + | + | + |
| Double humps of vertebral bodies | + | + | + | NA | + | NA | NA | + |
| Odontoid hyploplasia | + | NA | + | NA | + | NA | NA | − |
| Short and broad ilia with basilar hypoplasia | + | + | + | NA | NA | NA | NA | + |
| Lacy iliac crests | + | + | − | + | NA | NA | NA | + |
| Progressive acetabular slant and hip subluxation | + | + | + | NA | + | NA | NA | NA |
| Delayed ossification of proximal femoral epiphyses with horizontal growth plate | NA | NA | NA | NA | NA | NA | NA | + |
| Laterally displaced proximal femoral epiphyses | − | + | + | NA | NA | NA | NA | − |
| Epi-metaphyseal irregularities of long bones | Shoulder, elbow, knee | NA | Shoulder, elbow, knee, wrist | NA | NA | NA | NA | Hip |
| Brachydactyly with irregular epi-metaphyses of short tubular bones | + | + | + | NA | + | NA | NA | + |
| Delayed carpal ossification | + | + | + | NA | NA | NA | NA | NA |
| Mutations in RAB33B | c.211C>T | c.365T>C | c.48delCGGGGCAG and c.490C>T | c.136A>C | c.136A>C | c.136A>C | c.136A>C | c.444T>A |
- +, present; −, absent; NA, not available.
Mutation Analysis
Genomic DNA from all three patients was sequenced for the entire coding region as well as the splice sites of DYM gene. There was no significant sequence variation from the reference genome. Sequencing of RAB33B gene revealed homozygous novel sequence variants, c.211C>T (p.R71*) in exon one of patient one, c.365T>C (p.F122S) in exon two of patient two and compound heterozygous variants c.48delCGGGGCAG (p.G17Vfs*58) and c.490C>T (p.Q164*) in exons 1 and 2, respectively, of patient three (Fig. 5). The biallelic segregation was confirmed by sequencing parents’ samples in all the three families. The four variants reported here were not present in dbSNP, 1000 genome project or Exome Aggregation Consortium (ExAC) database. Conservation analysis of human RAB33B protein with other species showed that the variant c.365T>C resulted in the substitution of phenylalanine by serine at a highly conserved codon 122 (Fig. 6). This variant was also found to be damaging by pathogenicity prediction tools MutationTaster, SIFT, and PolyPhen-2. The other three variations were seen to be protein truncating mutations.


DISCUSSION
Genetic heterogeneity of SMC was previously suspected [Kaufman et al., 1971; Neumann et al., 2006]. Autozygosity mapping and exome sequencing in a large consanguineous Saudi family with SMC revealed a locus on 4q31.1 and missense mutations in RAB33B in the family and the other consanguineous Turkish family, respectively. RAB33B has two exons and it encodes for the RAB33B protein which belongs to a small GTP-binding family. This protein is localized to the Golgi apparatus and is involved in Golgi trafficking like DYMECLIN protein that is coded by DYM [Zheng et al., 1998; Starr et al., 2010; Alshammari et al., 2012]. The missense mutation, c.136A>C (p.K46Q) in exon 1 found in the Saudi family caused striking reduction in the protein probably due to its instability [Alshammari et al., 2012]. The mutation, c.444T>A (p.N148K) in exon 2 in the Turkish family also gave rise to marked reduction in the RAB33B protein [Dupuis et al., 2013].
In this report, all three patients exhibited typical clinical and radiological features of SMC. Common clinical features include short trunk, barrel chest, limited extension of elbow joints and genu valgum. Radiographs show platyspondyly with double humps of vertebral bodies, short and broad ilia with basilar hypoplasia and lacy iliac crests. All these features are in concordance with the previous reports [Neumann et al., 2006; Alshammari et al., 2012; Dupuis et al., 2013]. The phenotype hence appears homogeneous regardless of mutations in RAB33B and DMC. The three patients harbored four novel pathogenic variants: (c.211C>T (p.R71*), c.365T>C (p.F122S), c.48delCGGGGCAG (p.G17Vfs*58), and c.490C>T (p.Q164*)). Of the four pathogenic variants, two are novel nonsense mutations c.211C>T and c.490C>T, resulting in premature termination of translation at residues 71 and 164, respectively. The pathogenic variant, c.48delCGGGGCAG is a novel frame-shift mutation resulting in termination of translation at residues 58. The pathogenicity prediction by the in silico prediction tools as well as the multiple sequence alignment showed the variant, c.365T>C to be pathogenic and amino acid to be positioned in a highly conserved region.
In conclusion, the present study describes the clinical and radiological profile of three additional patients with SMC due to four novel mutations in RAB33B.
ACKNOWLEDGMENTS
The authors thank the families for cooperating in this study. KMG has received funds from Department of Science and Technology for the project titled “Application of autozygosity mapping and exome sequencing to identify genetic basis of disorders of skeletal development” (SB/SO/HS/005/2014).