Partially methylated alleles, microdeletion, and tissue mosaicism in a fragile X male with tremor and ataxia at 30 years of age: A case report
Abstract
CGG repeat expansion >200 within FMR1, termed full mutation (FM), has been associated with promoter methylation, consequent silencing of gene expression and fragile X syndrome (FXS)—a common cause of intellectual disability and co-morbid autism. Unmethylated premutation (55–199 repeats) and FM alleles have been associated with fragile X related tremor/ataxia syndrome (FXTAS), a late onset neurodegenerative disorder. Here we present a 33-year-old male with FXS, with white matter changes and progressive deterioration in gait with cerebellar signs consistent with probable FXTAS; there was no evidence of any other cerebellar pathology. We show that he has tissue mosaicism in blood, saliva, and buccal samples for the size and methylation of his expanded alleles and a de novo, unmethylated microdeletion. This microdeletion involves a ∼80 bp sequence in the FMR1 promoter as well as complete loss of the CGG repeat in a proportion of cells. Despite FMR1 mRNA levels in blood within the normal range, the methylation and CGG sizing results are consistent with the diagnosis of concurrent FXS and probable FXTAS. The demonstrated presence of unmethylated FM alleles would explain the manifestation of milder than expected cognitive and behavioral impairments and early onset of cerebellar ataxia. Our case suggests that individuals with FXS, who manifest symptoms of FXTAS, may benefit from more detailed laboratory testing. © 2016 Wiley Periodicals, Inc.
INTRODUCTION
Fragile X syndrome (FXS) is the most common heritable form of intellectual disability and autism, found in 1 in 4,000 in males and 1 in 8,000 in females in the general population, reviewed in [Hagerman et al., 2009]. FXS usually results from CGG repeat expansion >200, termed “full mutation” (FM), subsequent methylation of the regulatory regions and silencing of the FMR1 gene located on the X chromosome [Godler et al., 2010]. Fragile X associated tremor/ataxia syndrome (FXTAS) is a “late-onset” neurodegenerative disorder affecting carriers of the CGG expansion between 55 and 199 repeats, termed premutation (PM) [Jacquemont et al., 2004]. These PM alleles have estimated frequencies of 1 in 468 males and 1 in 151 females in the general population, and are associated with over-expression of the FMR1 gene and toxicity from excessive mRNA production leading to cell death contributing to FXTAS pathology, reviewed in [Hagerman et al., 2009]. Probable FXTAS has also been described in FM males with an unmethylated FMR1 promoter region (UFM), resulting in active FMR1 gene and IQ greater than 70 [Loesch et al., 2012; Santa Maria et al., 2013]. Here we present a man with FXS and clinical features consistent with probable FXTAS first manifesting in his early 30s. We show that he has unmethylated as well as methylated FM alleles, tissue mosaicism for the size of his expanded FMR1 alleles and a de novo microdeletion in the FMR1 promoter involving complete loss of the CGG expansion.
CLINICAL REPORT
Clinical History and Sample Processing
At 5 years of age, this male was diagnosed with FXS based on morphological features and cytogenetic testing showing 40% of cells expressing the Xq27.3 fragile site. At 33 years of age he was referred to the neurology service for assessment of progressive unsteadiness developing over 18 months. There was no accompanying weakness or sensory disturbance. His physical examination revealed mildly increased tone in all limbs, no tremor, mild symmetrical proximal weakness in the lower limbs, and moderate midline ataxia.
Twenty milliliters of peripheral blood was collected in EDTA tubes for CGG sizing, methylation, and FMR1 mRNA analyses. Buccal saliva samples were also collected using the Master Amp Buccal Swab Brush kit (Epicentre Technologies, Chicago, IL) and Oragene DNA OG-500 saliva collection kit (Genotek, Ottawa, Canada), respectively. DNA was extracted from 15 ml of blood and from buccal brush samples using the NucleoSpin®Tissue genomic DNA extraction kit (Machery–Nagel, Düren, Germany). Five milliliters of blood was used for peripheral blood mononuclear cell (PBMC) isolation using Ficoll gradient separation [Loesch et al., 2011]. RNA was extracted from the isolated PBMCs using RNeasy kit (Qiagen Global, Hilden, Germany) as per the manufacturer's instructions.
FMR1 Methylation, mRNA, CGG, and Proximal Region Sizing/Copy Number Changes
CGG repeat sizing in all samples was performed using polymerase chain reaction (PCR) as previously described [Khaniani et al., 2008]. The AmplideX™ FMR1 PCR Kit was used to perform CGG sizing on and determine mean methylation at two HpaII sites, 5′ and 3′ of the CGG expansion using blood DNA as per manufacturer's instructions (Asuragen, Austin, TX). Methylation sensitive Southern blot was used to determine CGG size and methylation of the FMR1 CpG island 5′ of the CGG expansion. Appropriate normal CGG size, PM and FM DNA samples used in a previous publication [Godler et al., 2010] were included as methylation controls. Seven to nine microgram of DNA were digested with either methylation sensitive enzyme combination of NruI/HindIII or EagI/ EcoRI or PstI which is not sensitive to methylation changes (Fig. 1). P32 labelled Pfxa3a probe (PCR Dig Synthesis kit; Roche Diagnostics Global, Basel, Switzerland) was used to detect FMR1 alleles by autoradiography as previously described [Godler et al., 2010], with the PS8 control probe used for the PstI digestion.

FREE1 and FREE2 PCR products were synthesized using primers and conditions previously described [Godler et al., 2010], and sized using capillary electrophoresis to rule out presence of the deletion at the 5′ epigenetic boundary of the FMR1 promoter and the exon1/intron 1 boundary at the 3′ end of the FMR1 promoter (Fig. 1). The Sequenom MALDI-TOF MS EpiTYPER system (Sequenom, San Diego, CA) was used to analyse mean methylation of the FREE2 region located 3′ of the CGG repeat in blood, buccal, and saliva samples, as previously described [Godler et al., 2010]. Two and a half million probe based microarray analysis was performed using blood DNA, to rule out large pathogenic copy number changes other than within FMR1, as well as to examine copy number changes of SNPs proximal to the CGG expansion, as previously described [Motoike et al., 2014]. FMR1 mRNA was reverse-transcribed in two separate cDNA reactions and each cDNA sample was analysed using real time-PCR reactions, as previously described [Loesch et al., 2011].
Clinical Assessments
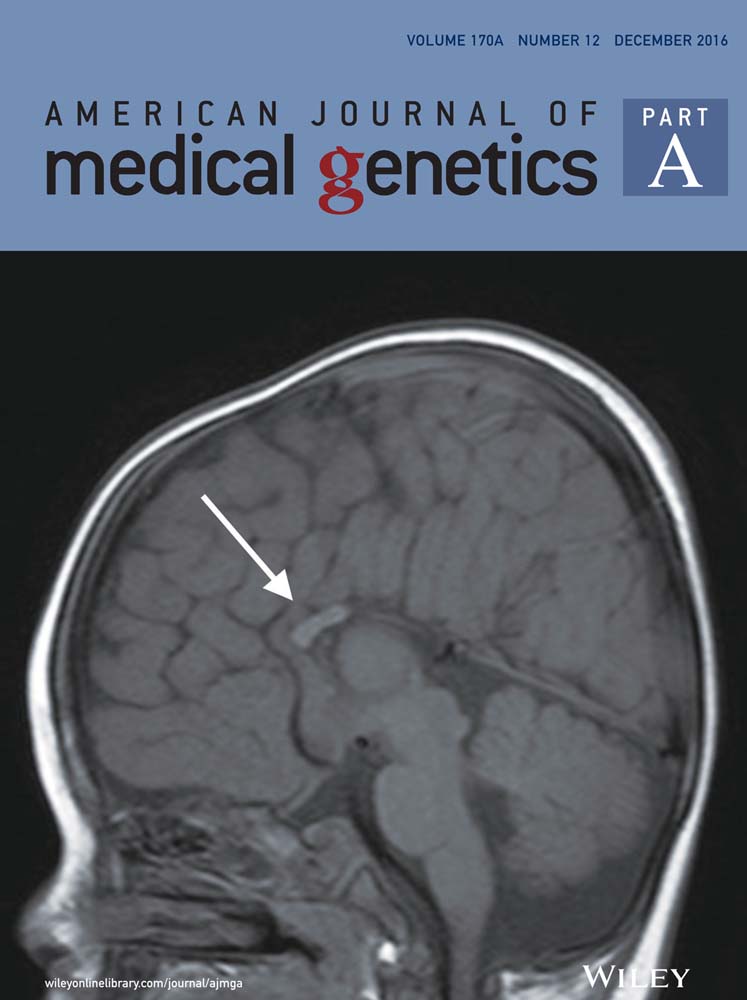
Blood tests, including thyroid function tests, vitamin B12, and autoimmune antibody panel, were within normal limits. Nerve conduction studies performed at the time did not reveal any evidence of peripheral neuropathy. MRI of his brain and spine revealed normal cerebellum without white matter lesions in the middle cerebellar peduncles and diffuse but non-specific T2 hyperintensities in the supratentorial cerebral white matter (Fig. 2) that were stable on repeat imaging 9 months later despite development of spastic paraparesis in the intervening period. These radiological findings and the gait ataxia met the diagnostic criteria for probable FXTAS [Loesch et al., 2005]. He was referred to a clinical geneticist to explore the possible presence of PM and unmethylated FM alleles.

At 35 years of age formal psychological assessment for this male was also performed. The Wechsler Adult Intelligence Scale (WAIS-IV) [Wechsler, 2008] showed that his cognitive ability was in the “Extremely Low” range with a full scale IQ of 51 (percentile rank 0.1; 95% confidence interval: 48–56). His WAIS-IV indexes scores did not differ significantly and were all within the “Extremely Low” range. The Autism Diagnostic Observation Schedule—Second Edition (ADOS-2) [Lord et al., 2012] assessment revealed some behavioral features consistent with autism spectrum disorder including deficits in communication skills, reciprocal social interaction, and social-emotional reciprocity that appeared to be separate from his impairment in verbal communication and general intellectual function. However, he did not satisfy the diagnostic criteria for autism spectrum disorder as per Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (American Psychiatric Association, 2013).
Testing for Causes of Ataxia
Friedreich ataxia (FA) and Spinocerebellar Ataxias (SCA) repeat regions were examined using PCR and gel electrophoresis, as previously described as in previously described protocols [Campuzano et al., 1996; Biros and Forrest, 1998]. Furthermore, omni2.5-8 microarray (Illumina, San Diego, CA) analysis did not identify copy number changes associated with whole exon deletions/duplications in the known ataxia or neuropathy genes, based on average probe density [Motoike et al., 2014].
RESULTS AND DISCUSSION
CGG Sizing and Detection of Microdeletion
Southern blot FMR1 CpG island methylation analysis in blood showed unmethylated fragments between 2.5 and 4.5 Kb for NruI/HindIII digestion and 2.6–4.4 Kb for EagI/EcoRI digestion (Fig. 1A). There were also methylated fragments between 4.7 and 6.5 Kb for NruI/HindIII digestion and 5.2 and 6.1 Kb for EagI/EcoRI digestion. For the PstI digestion (that was not methylation sensitive) the expanded alleles ranged in size from 1.63 to 2.83 Kb. Together the two methylation sensitive digestions, NruI/HindIII and EagI/EcoRI, suggested presence of unmethylated and methylated FM alleles ranging in size from 463 to 846 CGG repeats (Fig. 1A and B). The presence of FM alleles within this size range was further confirmed by the PstI digestion based Southern blot in buccal DNA (Fig. 1C).
All three digests followed by Southern blot also detected a fragment that was ∼0.2 Kb smaller than that in normal controls. This smaller allele was exclusively unmethylated and absent from the patient's mother's blood, the mother herself being a mosaic of PM and FM alleles (Fig. 1C). The absence of the smaller allele in the mother suggested that the pro-band is mosaic for a de novo microdeletion and FM expansion. As the same smaller band was detected in all three digests, a microdeletion would have to be located within PstI/PstI fragment and also within the overlapping NruI/HindIII and EagI/EcoRI fragments (Fig. 1A). Within this PstI fragment, the microdeletion is unlikely to occur within the exon 1 or intron 1 of FMR1 as microarray SNP copy number proximal to the repeat within intron 1 and PCR amplicon sizing of the FREE2 region on the exon1/intron 1 boundary were both completely normal (Fig. 1A and D). In this context, it is important to note that microarray SNP analysis did not identify any pathogenic variants elsewhere in the genome and the amplicon size of FREE1 located at the 5′ end of the FMR1 promoter was also completely normal (Fig. 1D). This microdeletion must therefore, involve complete loss of the CGG expansion together with a proximal binding site of one of the AmplideX PCR primers as the microdeletion was not detected using AmplideX PCR (Fig. 1E). Furthermore, this microdeletion must include loss of the AmplideX primer binding site located within the CpG island located ∼87 bp 5′ of the CGG repeat, as in a previous case [Schmucker et al., 1996].
Discordance Between Southern Blot and AmplideX PCR CGG Sizing
Unmethylated PM alleles of ∼70 repeats were detected by Southern blot following NruI/HindIII and EagI/EcoRI digests, as indicated by the lower points of smears of ∼2.9 and 3.0 Kb in size, respectively (Fig 1B). However, PM alleles were not detected using AmplideX PCR or standard sizing PCR [Khaniani et al., 2008], the latter having primer binding sites proximal to those of AmplideX. Only partially methylated FM size alleles were detected using AmplideX PCR. This discordance is unlikely to be due to the sizing imprecision of our Southern blot analyses (±30 CGG repeats) as an extended Southern blot smear is still within the PM range. An alternative explanation could be microdeletion involving both a portion of CGG repeat and the AmplideX primer binding site, similar to the smaller ∼2.5 kb Southern blot band that was also undetected using AmplideX PCR.
Discordance Between Southern Blot and Amplidex PCR Methylation Analysis
Two prominent FM allele types were detected in blood, buccal, and saliva DNA using AmplideX PCR (Fig. 2E). These alleles were also identified by Southern blot after NruI/HindIII, EagI/EcoRI, and PstI digests. However, there was significant discordance between the methylation percentage estimated by Southern blot following NruI/HindIII and EagI/EcoRI digestion and that by AmplideX analysis of two HpaII sites located either side of the expansion (Fig. 1). The mean AmplideX HpaII methylation estimate was ∼60% in blood and saliva and 94% in buccal DNA (Fig. 1E). In contrast, NruI and EagI methylation values were only 11% and 12%, respectively for Southern blot (100× density of all methylated bands/total of unmethylated and methylated bands) (Fig. 1B). If the density of the ∼2.5 Kb microdeletion band is not taken out of this calculation (as AmplideX does not detect it), NruI and EagI methylation values increase to 28% and 26%, respectively. This discordance could be because sizing and methylation analysis of all alleles that also have the microdeletion with the AmlideX primer binding sites (exclusively unmethylated by southern blot) being missed, leading to over-estimation of methylation by AmplideX for this case.
FREE2 Methylation, FMR1 mRNA Analyses and Testing for Other Genetic Causes of Ataxia
Increase in FREE2 methylation has been previously linked to FMR1 protein (FMRP) deficiency and the severity of cognitive impairment in FM males and females with FXS [Godler et al., 2012]. In this male, blood and saliva showed mean FREE2 methylation output ratio (MOR) of ∼0.4, while buccal samples showed MOR of ∼0.45 (Fig. 2C). Interestingly, the methylation values remained un-changed between blood and saliva samples collected 1.5 years apart. All MOR values were above those of normal allele and PM methylation ranges in males, but were below the FREE2 methylation range for typical FXS males carrying only FM alleles. These MOR values suggest that approximately 40% of leukocytes and ∼45% of epithelial (buccal) cells have methylated alleles. This is consistent with the patient's milder cognitive and behavioral impairments compared to those of “typical FXS” males carrying only highly methylated FM alleles. While unmethylated FM alleles have been associated with overexpression of FMR1 mRNA and increased risk of FXTAS [Loesch et al., 2012], it is not clear if the PM alleles with the proposed microdeletion could also contribute to the probable FXTAS phenotype observed in this patient (Fig. 1).
Analysis of FMR1 transcription in PBMCs revealed an mRNA level of 0.97 arbitrary units which is within the normal male expression range, above the levels observed for FM only and PM/FM mosaic males with the typical FXS phenotype (Fig. 2D). This is consistent with FREE2 methylation results where more than half of all cells would have an unmethylated promoter, overexpress FMR1 at twice the normal level, as previously described [Loesch et al., 2011, 2012], but overall would show normal FMR1 mRNA levels because the remaining cells would have a completely methylated FMR1 promoter with no FMR1 mRNA expression. Testing for other causes of ataxia excluded Friedrich ataxia, spinocerebellar ataxias type 1, 2, 3, 6, and 7 by PCR; and copy number changes associated with common peripheral neuropathies such as CMT1A and HNPP by the genome-wide array testing.
CONCLUSION
This is the first reported case of an FXS male showing white matter changes and progressive deterioration in gait with cerebellar signs, meeting the diagnostic criteria of probable FXTAS at 30 years of age. Our patient has cerebellar dysfunction with no evidence of any other cerebellar pathology and has molecular features of mosaicism for CGG repeat size, a de-novo microduplication within the FMR1 promoter for a proportion of cells, methylation mosaicism amongst different CpG sites and tissue types and UFM alleles that are consistent with FXTAS clinically manifesting concurrently with FXS. Our case adds to the expanding and evolving clinical and genetic profile of FXTAS and FXS, suggests that assessment for FXTAS may need to be broadened to detect males with partially methylated FM alleles and highlights the need for comprehensive testing to fully describe the range of underlying molecular pathologies.
AUTHORS’ CONTRIBUTIONS
YH, CR, MH, and RH clinically assessed the patient. MA and LB performed the psychological tests mentioned in the paper. DEG, DF, XL, AS, and HS performed the molecular analyses related to FXS and FXTAS including Asuragen PCR assay. BC performed the tests for other causes of inherited ataxias. YH and DEG prepared the drafts of the paper. All authors have reviewed, provided corrections and agree with the submitted version of the paper.
ACKNOWLEDGMENTS
This work was supported by the Victorian Government's Operational Infrastructure Support Program, Murdoch Childrens Research Institute, Royal Children's Hospital Foundation, NHMRC development grant [No 1017263 to DEG], Pierce Armstrong Trust [to DEG], NHMRC project grant [No 104299 to DEG.]. We thank the study participant and his family for their contribution and Dr. Benjamin Ong from the Sequenom Platform Facility (MCRI).