Whole exome sequencing is necessary to clarify ID/DD cases with de novo copy number variants of uncertain significance: Two proof-of-concept examples
Abstract
Whole exome sequencing (WES) is a powerful tool to identify clinically undefined forms of intellectual disability/developmental delay (ID/DD), especially in consanguineous families. Here we report the genetic definition of two sporadic cases, with syndromic ID/DD for whom array—Comparative Genomic Hybridization (aCGH) identified a de novo copy number variant (CNV) of uncertain significance. The phenotypes included microcephaly with brachycephaly and a distinctive facies in one proband, and hypotonia in the legs and mild ataxia in the other. WES allowed identification of a functionally relevant homozygous variant affecting a known disease gene for rare syndromic ID/DD in each proband, that is, c.1423C>T (p.Arg377*) in the Trafficking Protein Particle Complex 9 (TRAPPC9), and c.154T>C (p.Cys52Arg) in the Very Low Density Lipoprotein Receptor (VLDLR). Four mutations affecting TRAPPC9 have been previously reported, and the present finding further depicts this syndromic form of ID, which includes microcephaly with brachycephaly, corpus callosum hypoplasia, facial dysmorphism, and overweight. VLDLR-associated cerebellar hypoplasia (VLDLR-CH) is characterized by non-progressive congenital ataxia and moderate-to-profound intellectual disability. The c.154T>C (p.Cys52Arg) mutation was associated with a very mild form of ataxia, mild intellectual disability, and cerebellar hypoplasia without cortical gyri simplification. In conclusion, we report two novel cases with rare causes of autosomal recessive ID, which document how interpreting de novo array-CGH variants represents a challenge in consanguineous families; as such, clinical WES should be considered in diagnostic testing. © 2016 Wiley Periodicals, Inc.
INTRODUCTION
Array-CGH is a widely used technology, that is recommended as a first-tier test for postnatal evaluation of individuals with intellectual disability/developmental delay (ID/DD), autism spectrum disorders (ASD), and/or multiple congenital anomalies (MCA) [Manning et al., 2010; Miller et al., 2010]. Pathogenic variants are detected in 15–20% of ID/DD patients [Vissers et al., 2010b], who generally carry a deletion/duplication involving a known disease-associated genomic region or spanning one or more disease genes. As the identification of unreported copy number variants (CNVs) can be challenging to interpret, the American College of Medical Genetics (ACMG) has developed guidelines for reporting these CNVs [Kearney et al., 2011]. Rearrangements should be listed as benign or pathogenic, or reported as variants of unknown clinical significance. This latter category being fairly broad and including findings clearly demonstrated subsequently to be either pathogenic or benign.
Important recommendations to evaluate, and clinically interpret a specific CNV include whether it comprises gene-rich regions or is devoid of genes as well as the type of genes involved. Notably, the de novo nature of a CNV has been considered to be an important indication of its involvement in neurodevelopmental and neuropsychiatric disorders [Sebat et al., 2007; Pinto et al., 2010; Levy et al., 2011; Sanders et al., 2011]. Other associations, including the higher prevalence of de novo variants reported in sporadic schizophrenia cases compared to controls (10% vs. 1.3%) [Xu et al., 2012, 2008], also support this interpretation.
Here, we report two consanguineous families with probands exhibiting sporadic syndromic ID/DD for whom a de novo CNV required interpretation. In both cases, whole exome sequencing (WES) was crucial for a correct diagnosis, allowing identification of the disease-causing mutations, and thus, each CNV was reconsidered as not being the causative event underlying the disorder.
MATERIALS AND METHODS
Clinical Report
In our survey of over 900 patients with ID/DD or multiple congenital anomalies referred for array-CGH diagnostic screening from 2008 to 2014, we identified two patients who were born to consanguineous parents having a de novo CNV. Our study was performed with the approval of the Internal Review Board, and informed consent forms were obtained by the patients’ legal representatives.
Patient 296553
Patient 296553 was a 4-year-old girl born after an uneventful pregnancy. Parents were second cousins of Egyptian origin. She was referred to the pediatric genetics unit for severe developmental delay. Upon physical examination, the subject displayed microcephaly with brachycephaly (OFC 45 cm, <3rd centile) and a distinctive facies characterized by a round face, thin and horizontal eyebrows, synophrys, deep set eyes, a wide nasal bridge, and a thin upper lip (Fig. 1A and B). She could walk with support; speech was absent, and stereotypic movements were apparent (hand shaking, waving, and body rocking). Brain magnetic resonance imaging (MRI) performed at 3 years showed severe corpus callosum thinning (Fig. 1C) and a clear reduction of the white matter with poor myelination (Fig. 1C–E); the cerebellum was normal (Fig. 1C and D). Independent walking was achieved at the age of 5 years. At 7-year-old (last examination), the parents complained of frequent nocturnal awakenings and temper tantrums with self-injury; weight was 30 kg (97th centile), height 120 cm (50th centile), OFC 47 cm (<3rd centile). She presented with severe ID, language limited to a few syllabi and motor stereotypies.

Patient 296528
Patient 296528 was the second child of Moroccan origin first cousins. Family history was remarkable for a first cousin affected by severe ID (independent walking achieved at 10 years) and strabismus. Pregnancy was reported normal. She was born at 39 weeks of gestation with normal auxometric parameters (weight: 3,560 gr; length: 49 cm; occipito-frontal circumference (OFC): 35 cm), APGAR scores were 9/9. Global developmental delay was diagnosed at the age of 2 years, when she achieved independent ambulation. At that time, neurological evaluation disclosed hypotonia in the legs and mild ataxia. The patient was therefore referred for pediatric genetic evaluation, where she was recorded as having a weight and height in the 25th centile, ataxic wide-based ambulation, bilateral pes planus, difficulties in subtle manipulation; facial dysmorphisms were not apparent (Fig. 1F and G). Brain MRI detected severe cerebellar vermis hypoplasia with enlarged brain cerebrospinal fluid spaces. Cortical gyration was normal (Fig. 1H–J). Further investigations, including electroencephalography, ophthalmological evaluation, and general and metabolic workup (blood count, creatine phosphokinase [CPK], lipid profile, serum albumin, liver enzymes, transferrin, lactate, plasma acylcarnitine, transferrin isoelectrofocusing, and Vitamin E) did not provide informative data for diagnosis. At the age of 6 years (last evaluation), the height was 107 cm (10th centile), OFC 50 cm (25th centile); gait ataxia was regressed, and the patient walked independently without aid. Mild dysmetria was present during finger-nose and heel-shin tests. Dysarthria was present. Ophthalmological examination was normal.
Karyotyping and Array-CGH Analyses
Karyotyping was performed on GTG-banded chromosomes from circulating leukocytes. Paternity was confirmed by microsatellite analyses.
Array-CGH was performed using a 60 K whole-genome oligonucleotide microarray following the manufacturer's protocol (Agilent Technologies, Santa Clara, CA). Slides were scanned using a G2565BA scanner, and analyzed using Agilent CGH Analytics software v. 4.0.81 (Agilent Technologies) with the statistical algorithm ADM-2 and a sensitivity threshold of 6.0. Significant copy-number changes were identified by at least three consecutive aberrant probes. Reference human genomic DNA was GRCh37/hg19. Real-time PCR was used to confirm the array-CGH data and to further define the rearrangements (Suppl. Fig. S1). Patients were then submitted to the Decipher database (ID codes 296553 and 296528; https://decipher.sanger.ac.uk).
WES Analysis
WES was outsourced to BGI-Shenzen using genomic DNA extracted from circulating leukocytes. Targeted enrichment was performed using Nimblegen SeqCap EZ Library v.3.0 (64 M) (Roche Diagnostics, Mannheim, Germany), and captured libraries were loaded onto an Illumina HiSeq 2000 platform (Illumina, San Diego, CA). WES data analysis was performed using an in-house implemented pipeline [Cordeddu et al., 2014; Kortum et al., 2015; Niceta et al., 2015]. In brief, paired-end reads were aligned to the human genome (UCSC GRCh37/hg19) using the Burrows–Wheeler Aligner (BWA V. 0.7.5a-r405) [Li and Durbin, 2009], and presumed PCR duplicates were discarded using the Picard's MarkDuplicates utility (http://picard.sourceforge.net). The alignment process was refined by local realignment and base-quality-score recalibration steps by means of a Genome Analysis Toolkit (GATK 3.2) [McKenna et al., 2010]. GATK Unified Genotyper and Haplotype Caller were used to identify single nucleotide polymorphisms (SNPs) and insertions/deletions (INDELs) [DePristo et al., 2011]. Variants with a quality score <50 and quality-by-depth <1.5 or with results from four or more reads having ambiguous mapping (this number being >10% of all aligned reads) were discarded. Remaining variants were then filtered against the available public database (dbSNP141, retaining only variants with MAF <0.001 or with a known clinical association), and the in-house database (retaining variants with frequency <5%). SnpEff toolbox v3.6 [Cingolani et al., 2012] was used to predict the functional impact of variants, and retain missense/nonsense/frameshift changes, coding indels, and intronic variants at exon-intron junctions (within position −5/+5). Functional annotation of variants was performed by using snpEff v3.6 and dbNSFP2.8 [Cingolani et al., 2012; Liu et al., 2013].
Based on consanguinity, we assumed an autosomal recessive model of inheritance for both traits, and retained all the homozygous variants located within Loss of Heterozygosity (LoH) genomic stretches using the Homozygosity Mapper [Seelow and Schuelke, 2012] (http://www.homozygositymapper.org/), setting 35 as the number of consecutive homozygous SNPs. Retained variants were prioritized according to their predicted functional impact (SVM radial score >0 or CADD score >15) [Liu et al., 2013; Kircher et al., 2014], and their biological and clinical relevance.
Sequence validation and segregation analyses were performed by Sanger sequencing using an ABI 3130XL and the ABI BigDye Terminator Sequencing Kit V.3.1 (Life Technologies, Carlsbad, CA). Sequences were examined using the SeqScape v2.6 Software (Life Technologies).
RESULTS
Array-CGH
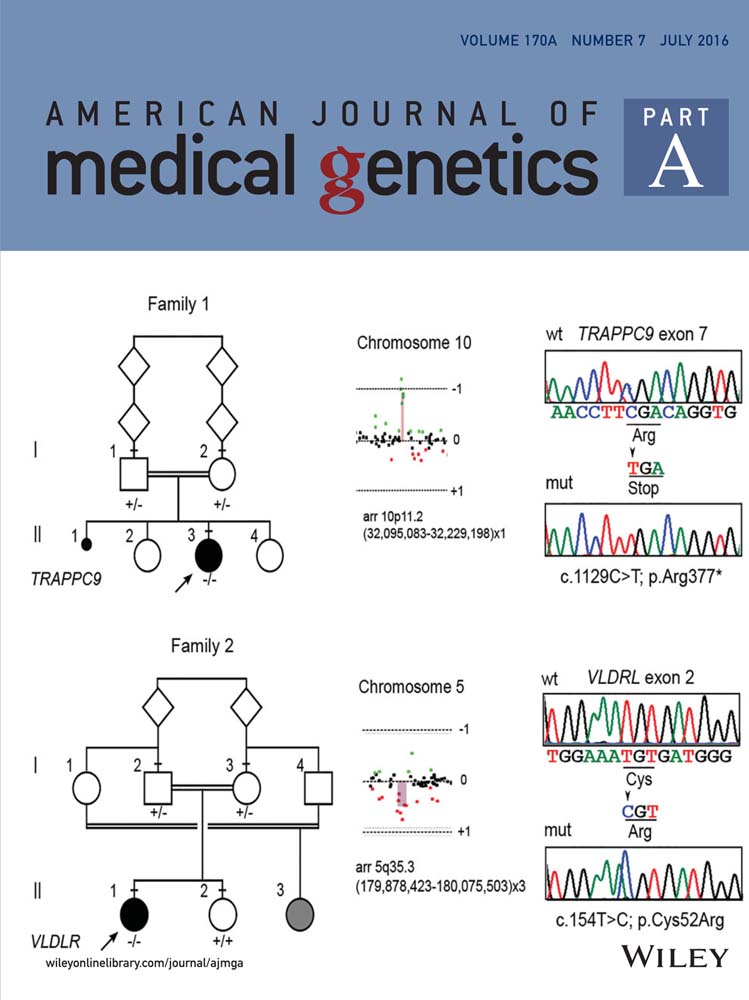
Array-CGH analysis documented a de novo 134–483 kb deletion on 10p11.22 in patient 296553 [arr 10p11.22(31,817,746x2,32,095,083-32,229,198x1,32,300,151x2)), hg19] spanning the ARHGAP12 gene (MIM 610577), and a de novo 200–345 kb duplication on 5q35.3 in patient 296528 [arr 5q35.3(179,807,078x2, 179,878,423-180,075,503x3,180,152,402x2), hg19] encompassing the CNOT6 (MIM 608951), SCGB3A1 (MIM 606500), and FLT4 (MIM 136352) genes (Figs. 2 and S1). Real-time PCR assays confirmed the rearrangements and their de novo origin, although we did not further define the limits of the duplicated genomic region in patient 296528 (Suppl. Fig. S1). The Decipher database reported three cases with a deletion and three with a duplication spanning ARHGAP12; all records referred to large rearrangements (3.5–10 Mb) encompassing multiple genes. Several rearrangements spanning CNOT6, SCGB3A1, and FLT4 are reported in the Decipher database, but all are large (>10 Mb), suggesting many genes may contribute to those phenotypes.

Exome Sequencing
WES statistics are reported in Supplemental Table SI. Data annotation predicted 12,859 (case 296553) and 12,476 (case 296528) high-quality variants having a functional impact (i.e., non-synonymous and splice site changes). Among them, 2,353 and 2,134 private, rare (minor allele frequency <0.001) or clinically associated changes were further analyzed. No variants were present in genes located within the identified de novo CNVs. Variants were filtered to retain rare or private homozygous sequence changes located within LoH regions, and in silico analyses of the predicted functional impact of individual variants and the biological relevance of the encoded proteins allowed identification of an excellent disease gene candidate in each patient (Suppl. Tables SII and SIII; Fig. S2). A nonsense change, c.1423C>T (chr8:141407724, hg19; p.Arg377*) (rs267607136, flagged as clinically associated), was identified in Trafficking Protein Particle Complex 9 (TRAPPC9, MIM 611966) in patient 296553 (Fig. 2). TRAPPC9 encodes a protein implicated in NF–kB activation, and five inactivating mutations in this gene have been reported to underlie a rare, recessive non-syndromic ID associated with microcephaly, mild cerebral white matter hypoplasia, and corpus callosum hypoplasia (MIM 613192) (Fig. 3), which matched with the clinical features exhibited by the proband.

Case 296528 was homozygous for a missense change, c.154T>C (chr9:2635524, hg19; p.Cys52Arg), in the Very Low Density Lipoprotein Receptor gene (VLDLR, MIM 192977) (Fig. 2). The affected residue is highly conserved (Suppl. Fig. S3), involved in an intramolecular disulfide bridge required for proper receptor function, and resides in the ligand-binding type repeat (LBTR) region. Consistently, substitution of this residue was predicted to be deleterious. Homozygous or compound heterozygous mutations in VLDLR have been reported to cause cerebellar ataxia, mental retardation, and disequilibrium syndrome type 1 (CAMRQ1; MIM 224050) (Fig. 3), a disorder with features that overlap those of our patient. In both probands, Sanger sequencing validated both sequence changes and segregation.
DISCUSSION
Guidelines for investigating the causality of unannotated CNVs consider their de novo origin as one of the most important factors [Lee et al., 2007; Buysse et al., 2009; Gijsbers et al., 2009; Koolen et al., 2009; Miller et al., 2010; Gijsbers et al., 2011]. Here we report two cases in whom array-CGH identified CNVs that were initially suspected to be causative of the disease because of their de novo occurrence in each proband. Patient 1 was found to carry a deletion encompassing ARGAPH12 encoding for a Rho GTPase-activating protein. Analogously to other proteins of the same family involved in ID (e.g., oligophrenin, OMIM 300486), ARHGAP12 haploinsufficiency was originally hypothesized to have a causative role in this patient. In the second patient, the duplicated region encompassed three protein-coding genes: FLT4, SCGB3A, and CNOT6. Given the role of transcription regulation in the pathogenesis of ID/DD [van Bokhoven, 2011], CNOT6, encoding the catalytic component of the CCR4-NOT core transcriptional regulation complex, was originally considered as being possibly causative for the disease, although it was classified as a variant of unknown significance.
Recent publications have reported that small de novo imbalances must not be automatically classified as causal for the investigated phenotype in the absence of strong evidence from other data sources, and rearrangements below 500 kb have to be considered carefully. A historical example of a de novo CNV being wrongly assigned as pathogenic is presented by the 250 kb deletion in MACROD2, which was described in a patient with Kabuki syndrome, later found to have a mutation in the MLL2 gene [Maas et al., 2007; Paulussen et al., 2011]. More recently, a de novo 86.5 kb deletion was reported as pathogenic in a patient with ID and eye disorder, because it harbored AMBRA1, a gene expressed in the neural retina and brain [Fimia et al., 2007]. Subsequent accurate clinical evaluation of the patient suggested a possible diagnosis within the clinical spectrum of CHARGE syndrome, which was confirmed by the identification of a CHD7 mutation, known to be causative of the disease [Vissers et al., 2004].
The patients in our study further support the caveats concerning small de novo CNVs, indicating the usefulness of WES in these cases, particularly when restricted to the scanning of genes that have been causally related to Mendelian disorders (i.e., the clinical exome). When complex phenotypes and traits are evident, without clinical diagnosis, the clinical exome should be considered in the presence of consanguinity or LoH detected by SNP-array, and in the particular cases in which array-CGH/SNP-array provides de novo structural events for which a clear pathogenicity is not provided. Indeed, in countries of the Mediterranean basin, the likelihood of reaching molecularly confirmed diagnosis in subjects from consanguineous families—having an unresolved but putatively autosomal-recessive disorder—has been estimated at around 36% using WES [Makrythanasis et al., 2014]. It should also be noted that WES has enabled an increase in the discovery rate of non-syndromic autosomal recessive ID (NS-ARID), identifying 32 new genes in the last 3 years [Musante and Ropers, 2014]. The number of genes causally associated with ID/DD is expected to continue to increase in the near future, which makes the clinical exome a particularly informative tool as WES data can be stored and made available for future investigations as new information emerges.
In this study, we report that WES analysis allowed identification of the causal molecular lesion in both patients. In the first family of Egyptian origin, a homozygous nonsense mutation (c.1423C>T; p.Arg377*) in TRAPPC9 was identified. TRAPPC9 has been implicated in NF–kB activation, and is possibly involved in intracellular trafficking. The same truncating lesion had been previously reported in families from Pakistan, Syria, and of Arab–Israeli origin [Mir et al., 2009; Mochida et al., 2009; Abou Jamra et al., 2011]. There are only five known mutations of this gene, all of which have a predicted inactivating effect (Fig. 3). The TRAPPC9 mutation-associated phenotype was initially reported as non-syndromic ID with postnatal microcephaly [Mir et al., 2009; Mochida et al., 2009; Philippe et al., 2009]. However, consistent with the present findings, more recent reports have provided evidence that loss of TRAPPC9 function underlies a syndromic form of ID with distinctive facial features (brachycephaly, round face, straight eyebrows, synophrys, deep set eyes, wide nasal bridge, and thin upper lip), true or relative microcephaly, MRI brain anomalies (corpus callosum hypoplasia, reduced white matter volume with multifocal hyperintensity), and overweight [Marangi et al., 2013]. Frequent sleep awakenings and motor stereotypies, also represent variably occurring features [Abou Jamra et al., 2011; Marangi et al., 2013].
In the second family, we identified a previously unreported homozygous missense change, c.154T > C (p.Cys52Arg) in the VLDLR gene. Analogously of the Low Density Lipoprotein Receptor (LDLR), the binding domain of VLDLR to lipoproteins contains seven tandem repeated cysteine-rich domains at its amino terminus [Fass et al., 1997] (Fig. 3). Each repeat of ∼40 amino acids contains two loops stabilized by three disulphide bridges, which are required for the correct folding of the domain. Cys52 is predicted to be involved in an intramolecular disulfide bond with Cys67 (http://www.uniprot.org/uniprot/P98155), and loss of this disulfide bridge is expected to result in protein misfolding and its degradation by the ER-associated protein degradation machinery (ERAD) [Ali et al., 2012]. Eleven mutations in this gene have been reported, most of which have a predicted loss-of-function mechanism (Fig. 3). Only three missense changes are known, all apparently associated with a classical CAMRQ1 phenotype. The clinical phenotype associated with VLDRL mutations is relatively homogeneous and includes non-progressive truncal ataxia, dysarthria, moderate-to-profound intellectual disability, and pes planus. MRI of patients with VLDRL mutations reveals cerebellar hypoplasia (mainly vermian) and a simplification of cortical gyri. Other symptoms, such as epilepsy, are variably associated with VLDRL mutations. Some mutations have been associated with quadrupedal locomotion [Tan, 2006; Ozcelik et al., 2008; Turkmen et al., 2008], although this was suggested to be a physical adaptation [Sonmez et al., 2013]. Notably, our patient exhibited a milder phenotype, which may be specifically associated with the type and location of mutation that could result in a receptor having an incompletely impaired function (see Fig. 3). MRI showed hypoplasia of cerebellar vermis, but cerebral gyration was normal, in contrast with all other reported cases.
In conclusion, diagnosis in both patients would have been missed or misled if solely based on the array-CGH data interpretation. Conversely, a SNP-based array would have been more informative in simultaneously searching for pathogenic CNVs and testing the presence of homozygosity regions, which are a signature for consanguinity.
This report further emphasizes the utility of WES to explore the possible occurrence of rare genetic disorders in consanguineous families even when de novo CNVs are found. To avoid misinterpretations, WES should be used together with array-CGH/SNP-based array as a first-tier diagnostic tool in consanguineous cases [Vissers et al., 2010a].
ACKNOWLEDGMENTS
We are grateful to all family members who contributed to this study. Our work was funded by MURST 60% (to A. Brusco), Ospedale Pediatrico Bambino Gesù (GeneRare to M.T.). We thank CINECA for computational resources (WES data analysis). This study makes use of data generated by the DECIPHER Consortium. A full list of centers which contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from [email protected]. Funding for the project was provided by the Wellcome Trust.