Spectrum of gene variants linked to cystic fibrosis in nonwhites
Research finds certain screening panels may be inadequate
Nonwhite patients with cystic fibrosis (CF) often have genetic mutations that are not included in a recommended carrier screening panel, according to recent research.
Caused by mutations in each of the two copies of the CFTR gene, CF involves production of abnormally thick, sticky mucus in the lungs and digestive system. Patients also have a spectrum of other CF- related symptoms—especially frequent respiratory infections, digestive issues, and growth problems in childhood—to varying degrees.
The disease is most commonly seen among people of non-Hispanic European ancestry, occurring in approximately 1 in 2,500 Northern-European whites and more commonly in Ashkenazi Jews. It is generally much less prevalent in nonwhite populations, but in some groups, such as African-Americans, CF is as common as some diseases on panels designed for carrier screening in Ashkenazi Jews.
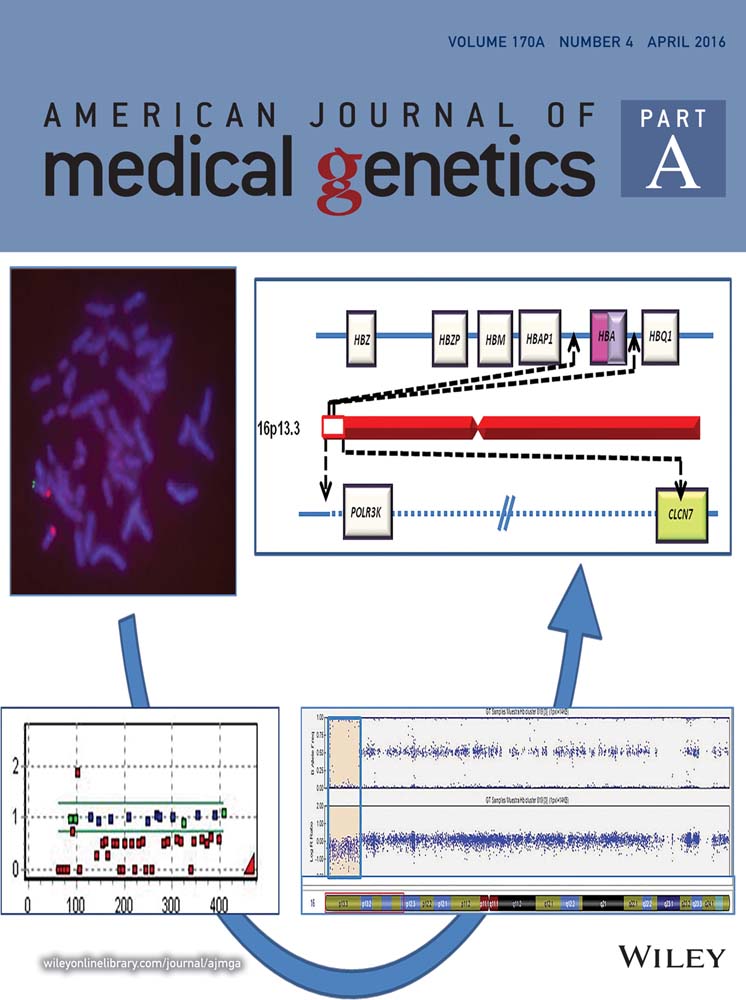
Writing in The Journal of Molecular Diagnostics, Stanford University–based researchers say that patients with CF of Hispanic, African-American, or Asian ancestry are less likely than whites to have CFTR variants included among the 23 variants recommended in carrier screening panels by the American College of Medical Genetics and Genomics (ACMG). Nonwhite patients with CF are more likely to have rare variants, plus duplications and deletions that diagnostic CFTR sequencing often doesn't detect (Schrijver et al., 2016).
“In California, every second baby born is of Hispanic origin, and nationally, ethnicities are increasingly diverse and mixed. So it is indeed important to have a good grasp of the diversity of the mutations in all populations in order to optimally serve our patients,” says study first author Iris Schrijver, MD, Professor of Pathology and Pediatrics at Stanford University Medical Center in California.
Nonwhite infants with cystic fibrosis may have rare genetic mutations that most states do not include in standard diagnostic or newborn screening panels.
The Study
The researchers began by identifying CFTR variants and their frequencies in nonwhite patients, then estimated the proportion of such patients who wouldn't be detected by the ACMG panel.
They identified a spectrum of CFTR variants among 140 ethnically diverse patients who had Sanger sequencing, which often misses large additions or deletions of base pairs in the DNA. If sequencing did not reveal two mutations in a patient, researchers also used multiplex ligation-dependent probe amplification (MLPA) to detect deletions and duplications involving one or more exons, which provide the genetic code for entire proteins.
Sanger sequencing identified two CFTR sequence variants in 89 patients. In the 51 who had MLPA testing, the researchers detected 14 deletions or duplications of various degrees of complexity in 12 patients. These included nine distinct variants, including six that occurred in nonwhites but had never been reported. These results show that MLPA should be applied routinely in diagnostic CFTR testing, researchers say.
How Common Are Variants?
The researchers then determined the type and frequency of genotypes by ethnicity of patients included in the Cystic Fibrosis Foundation Patient Registry between 2008 and 2013. It included 17,327 whites, 1,252 Hispanics, 558 African-Americans, 116 Native Americans, and 91 Asians.
Although previous studies have determined that the p.Phe508del variant is predominant in whites, the Schrijver et al. study found that registry patients from other groups were much more likely to carry other mutations. Overall, 10% of white patients carried no p.Phe508del mutation at all, compared to 40% of Asians, 38% of African-Americans, 30% of Hispanics, and 17% of Native Americans, and did not have this variant, the researchers write.
The ACMG panel, which was designed for carrier screening but is sometimes used for diagnosis and newborn screening in many states, would have determined both alleles for a majority of white and Native American patients in the registry but not for the majority of other ethnicities. The panel would not have provided complete genotyping for 69% of Asians, 62% of African-Americans, and 52% of Hispanics, the researchers write.
It is indeed important to have a good grasp of the diversity of the mutations in all populations in order to optimally serve our patients. — Dr. Schrijver
Reaction
Ruth Heim, PhD, applauds the paper's call for ethnic and racial equity in detection of disease-causing variants, noting it is in line with conclusions she has reached in her own research. However, “molecular diagnosis isn't the only way to go,” says Dr. Heim, Director of Clinical Operations at Integrated Genetics in Westborough, Massachusetts. The non-genetic test of the concentration of chloride in sweat is also diagnostic, she notes. Often, physicians familiar with CF can diagnose classic cases of the disease clinically. Molecular testing is more important in cases where symptoms aren't clear-cut, and it is necessary in newborns, she notes.
The paper didn't discuss some key issues, including whether patients' variants actually caused disease or how severe it was. Early diagnosis is more important for patients with more severe CF, Dr. Heim adds.
The researchers say their findings have implications for carrier and newborn screening as well as diagnostic testing. However, the director of molecular testing at a large academic reference lab identifies various molecular testing scenarios beyond the 23-variant panel.
“We use a panel that includes the ACMG 23 variants mainly as carrier screening but also as a first step for diagnostic testing,” says Elaine Lyon, PhD, Medical Director of Molecular Genetics/Genomics and also Co-Medical Director of Pharmacogenetics at ARUP Laboratories in Salt Lake City.
Many labs have larger panels. For example, Illumina's CF panel kit assay with 139 mutations is cleared by the U.S. Food and Drug Administration and can be used by other laboratories.
If the panel does not identify two causative mutations in patients with symptomatic CF, the next step is sequencing the gene. Some clinicians order it as a first step. If sequencing doesn't identify two causative mutations, testing should continue to detect deletions and duplications, says Dr. Lyon.
But physicians who are not very familiar with CF might not be aware of all of these testing options, notes MyMy Buu, MD, Clinical Assistant Professor of Pediatrics and Pediatric Pulmonology at Stanford School of Medicine in California. Her own research found that Hispanic patients with CF have a higher mortality rate than patients from other groups, even after adjusting for socioeconomic factors and disease severity.
Dr. Buu included patients who had been diagnosed with CF before California added the disease to its newborn screening panel. The state now includes a group of 40 CFTR mutations selected for its diverse population, she notes.
Dr. Lyon says the paper by Schrijver et al. offers a message that is consistent with good practice. “Look at the test offerings and make decisions that are best for individual patients. Most medical geneticists know this, but other specialists may not,” she adds.