Defining the phenotype associated with microduplication reciprocal to Sotos syndrome microdeletion
Abstract
NSD1 point mutations, submicroscopic deletions and intragenic deletions are the major cause of Sotos syndrome, characterized by pre-postnatal generalized overgrowth with advanced bone age, learning disability, seizures, distinctive facial phenotype. Reverse clinical phenotype due to 5q35 microduplication encompassing NSD1 gene has been reported so far in 27 cases presenting with delayed bone age, microcephaly, failure to thrive and seizures in some cases, further supporting a gene dosage effect of NSD1 on growth regulation and neurological functions. Here we depict the clinical presentation of three new cases with 5q35 microduplication outlining a novel syndrome characterized by microcephaly, short stature, developmental delay and in some cases delayed bone maturation, without any typical facial or osseous anomalies. © 2014 Wiley Periodicals, Inc.
INTRODUCTION
Sotos syndrome-1 [OMIM #117550] is a well-known genetic disease whose diagnosis is based on three cardinal features: typical facial gestalt, learning disability ranging from mild to severe, and pre-postnatal generalized overgrowth. Congenital heart defects, neonatal jaundice, renal malformations, seizures and behavioral problems are other features frequently present [Tatton-Brown et al., 2005]. Increased susceptibility to neoplasia such as neuroblastoma, acute lymphoblastic leukemia, and ganglioglioma is a still controversial feature [Lapunzina, 2005].
Recently, it has been shown that patients presenting with a Sotos-like phenotype, consisting of postnatal overgrowth, macrocephaly, advanced bone age, long narrow face, high forehead, slender habitus, scoliosis, unusual behavior with anxiety and intellectual disability may bear heterozygous mutation in the NFIX gene (OMIM: 164005) on chromosome 19p13.3 [Malan et al., 2010; Yoneda et al., 2012]. This condition has been defined as Sotos syndrome-2 (SOTOS2) [OMIM #614753]. Moreover, most cases of Sotos syndrome-2 are sporadic [Malan et al., 2010; Yoneda et al., 2012].
NSD1 (nuclear receptor SET domain containing protein-1, OMIM 606681) haploinsufficiency is the only known causative gene for Sotos syndrome-1. NSD1 protein presents a modular composition: two proline–tryptophan–tryptophan–proline (PWWP) domains, five canonical plant homeodomains (PHD1–PHD5), a variant C5HC3 PHD finger 6 (PHD6), and a SET domain that catalyzes methylation of histone H3 Lys36 (H3K36) or histone H4 Lys20 (H4K20) in vitro [Pasillas et al., 2011]. NSD1 regulates transcription of genes controlling normal differentiation. Although the mechanism by which NSD1 binds chromatin and regulates transcription is unknown, the fact that single Sotos mutations in the AWS/SET, PHD1, PHD2, PHD3, PHD4, PHD5, PHD6, and PWWP2 domains produce the same phenotype as 5q35 microdeletions that eliminate the entire NSD1 gene [Kurotaki et al., 2002; Cecconi et al., 2005] suggests that each of these domains plays an essential, non-redundant role [Pasillas et al., 2011]. Pasillas and colleagues suggested that point mutations in NSD1 PHD domains disrupt its transcriptional regulation by interfering with its ability to bind epigenetic marks and recruit cofactors [Pasillas et al., 2011].
The 1.9 Mb Sotos critical region on chromosome 5, containing approximately 21 genes (Fig. 1) is flanked by two well-characterized low-copy repeats (LCRs): a proximal 390 kb repeat (Sos-PREP) and a distal 429 kb repeat (Sos-DREP) [Kurotaki et al., 2005; Visser et al., 2005]. This genomic architecture makes the Sotos region prone to chromosomal rearrangements mediated by Non-Allelic Homologous Recombination (NAHR) so that a common 5q35 microdeletion mediated by directly oriented subunits within Sos-PREP and Sos-DREP is the most common mutational mechanism in the Japanese population associated with Sotos syndrome [Kurotaki et al., 2003], whereas intragenic mutations are responsible for at least 80% of Sotos patients from Europe and North American.

In general, for each deletion mediated by LCRs in NAHR, there is a reciprocal duplication. With the implementation of whole genome arrays, several duplications reciprocal to previously known microdeletion syndromes have been identified [Bolton et al., 2001; Somerville et al., 2005; Van Esch et al., 2005; Potocki et al., 2007; Ou et al., 2008; Torniero et al., 2008; Bi et al., 2009; Portnoi, 2009; Novara et al., 2010, 2013].
Duplications encompassing the NSD1 gene have been reported so far in 27 patients, including six familial cases (four from one family and two from another one) [Chen et al., 2006; Kirchhoff et al., 2007; Franco et al., 2010; Zhang et al., 2011; Dikow et al., 2013; Rosenfeld et al., 2013; Zilina et al., 2013] (Fig. 1). Most of the duplicated patients presented with short or low normal stature, microcephaly or low normal occipitofrontal head circumference, failure to thrive and seizures in some cases, further supporting a gene dosage effect of NSD1 on growth regulation and neurological functions [Chen et al., 2006; Kirchhoff et al., 2007; Franco et al., 2010; Zhang et al., 2011; Dikow et al., 2013; Rosenfeld et al., 2013; Zilina et al., 2013]. In 24 out of 27 cases, the duplication was mediated by Sos-PREP and Sos-DREP LCRs [Franco et al., 2010; Zhang et al., 2011; Dikow et al., 2013; Rosenfeld et al., 2013; Zilina et al., 2013].
Here we report on three new cases with 5q35 microduplications mediated by LCRs and identified by array-CGH. These cases help better define the phenotype of patients with microduplication reciprocal to Sotos deletion.
MATERIALS AND METHODS
Informed consent was obtained for all patients. Patient 1 did not give consent for her photos publication. Parents of Patients 2 and 3 gave permission for their photo publication.
Molecular Karyotyping
Molecular karyotyping was performed through array-CGH using the Agilent array 180 K for Patient 1 and 44 K for Patient 3 according to the manufacturer's protocol. Data analysis was performed using Agilent Genomic Workbench Standard Edition 6.5.0.58.
The Oxford Gene Technology Syndrome Plus v.2 2 × 105 K format was used for Patient 2. Labeling and hybridization was done according to standard protocol from Oxford Gene Technology and scanning was performed on a Perkin Elmer Scanarray gX scanner at a resolution of 5 μm. Genepix Pro 6.1 was used for feature extraction and Cytosure Interpret software v3.1.7 for data analysis.
Probe positions are referred to hg19.
QPCR
QPCR was performed in Patient 1 and her mother and brother. Specific target sequences were selected for Real-time quantitative PCR (qPCR) using Primer 3 software (http://frodo.wi.mit.edu/) and experiments were performed as previously described [Novara et al., 2013].
MLPA
MLPA was performed in Patient 2. The P064-B2 Mental Retardation-1 MLPA kit from MRC Holland was used for independent validation of the array-CGH findings and for investigation of parental blood samples. MLPA analysis was done according to standard protocol from MRC Holland. The P064 MLPA kit contains three probes for the NSD1 gene that are located into the region involved in deletion on chromosome 5q35.3.
FISH
FISH was performed on interphase nuclei from Patient 1, and on metaphase spreads of her mother and her brother in order to investigate if the duplication was direct or inverted and to highlight possible insertional translocations present in her relatives. To this aim, BAC clones allocated in the duplication region (RP11-627M5 and RP11-564G9) were used. DNAs extraction, labeling, hybridization, detection and FISH experiments were undertaken as previously described [Ciccone et al., 2005].
Patient 1
The proband, a Caucasian 46-year-old female, was the first child of nonconsanguineous healthy parents. No relatives with genetic diseases, malformations or multiple miscarriages/stillbirths were reported. No information about pregnancy and delivery were available. She attended schools for educationally subnormal children until 15 years of age. Menarche appeared at this age, menstrual cycles were regular except for an amenorrhea lasting 1 year. Starting from 25 years of age, she presented recurrent major depressive episodes but she did refuse to provide the neuropsychiatric records. No other relevant diseases were recorded.
The proband presented with mild intellectual deficiency (development milestones are not available). Clinical evaluation at 46 years of age showed: height 150 cm (−2.1 SD), weight 65 kg (+0.9 SD; WHO standard), head circumference 49.0 cm (−4.0 SD: severe microcephaly), thick eyebrows, short palpebral length (2.4 cm, −5.0 SD) and mild downslanting palpebral fissures. Inner canthal distance was 2.7 cm (−1.2 SD), interpupillary distance was 5.1 cm (−2.0 SD). Prominent nasal root and large nasal tip were present. Philtrum length was 1.0 cm (−2.0 SD), the upper lip vermilion was thin whereas the palate and the uvula were normal. Ear length was 5.9 cm (−0.4 SD). She presented long ear lobes, symmetrical round face, and retrognathia. Trapezius muscles were bilaterally prominent. Severe lumbar hyperlordosis, bilateral cubitus valgus, short hands with brachydactyly (palmar length: 8.7 cm, −3.7 SD) and middle finger length (6.5 cm, −3.6 SD) were present. She presented with ulnar deviation of both middle fingers (right > left) and radial deviation of both 4th fingers. Fifth fingers were very short and with a proximal implantation. She had limited thumbs adduction and bilateral thenar hypoplasia and broad big toes. Foot length was cm 21.5 (−1.7 SD) bilaterally. About 15–20 truncal melanocytic naevi were present. Moderate truncal obesity was also present (BMI 28.9, +1.5 SD).
Patient 2
The child (Decipher VEJ248956) was a 5-year-old female born to healthy nonconsanguineous parents. Birth weight was 2,900 g and birth length was 50 cm (gestation-age: 40 + 0; head circumference at birth: 34 cm; abdominal circumference: 31 cm; placenta weight: 530 g). At the age of 3 years and 6 months her length was 91.8 cm (−1.8 SD; WHO standard), weight 13.4 kg (−0.9 SD) and head circumference 49 cm (−0.01 SD). At last evaluation at 5 years and 2 months of age her length was 102.5 cm (−1.6 SD), weight 15.3 kg (−1.2 SD) and head circumference was not measured, because there was no obvious indication for microcephaly. Bone age has not been measured. She presented with a cleft palate, hearing problems, joint hypermobility, speech delay, and mild intellectual disability (speech test: 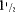 -year delay shown by speech test at age 4; intellectual disability: 3–4 months delay shown by Kuno-Beller test at age 9 months) (Fig. 2). She had growth retardation. Her hearing test right after birth was normal. She did not produce any sounds until age 1. She had delayed motor development and was very passive. No obvious dysmorphic features were present.
-year delay shown by speech test at age 4; intellectual disability: 3–4 months delay shown by Kuno-Beller test at age 9 months) (Fig. 2). She had growth retardation. Her hearing test right after birth was normal. She did not produce any sounds until age 1. She had delayed motor development and was very passive. No obvious dysmorphic features were present.

Patient 3
The proband is a 13-year-old boy referred because of short stature and borderline intellectual disability. Familial and prenatal history was unremarkable. He was born at 39 weeks by spontaneous delivery to nonconsanguineous parents; neonatal weight and length were 2,700 g (−1.63 SD) and 48 cm (−1.2 SD) respectively. Head circumference and APGAR scores were not reported. At birth, isolated severe hypospadias was observed.
The child walked unsupported at 2 years of age. He pronounced first words at 3 years. He attended a public school, but he experienced learning difficulties that required a special learning program. Parents refused any investigations to better define his IQ.
At clinical evaluation at 13 years of age, weight was 33 kg, height 138 cm and head circumference 51 cm, all at −2 SD. The facial dysmorphisms consisted in micro-brachycephaly and hypoplastic alae nasi with low-hanging columella (Fig. 2). Thyroid dysfunction, growth hormone deficiency and hypothalamic-pituitary axis dysfunction were ruled out by extensive endocrinological work-up that were performed to ruling out any metabolic causes of short stature. Bone age was severely delayed.
RESULTS
Molecular karyotyping performed by high-resolution array-CGH showed a 5q35.2q35.3 duplication of about 2.1, 2.08, and 1 Mb in size for Patients 1, 2, and 3 respectively. Unbalanced basepair positions were the following: arr 5q35.2q35.3(175,347,711–177,484,036) × 3 for Patient 1, arr 5q35.2q35.3(175,346,398–177,430,496) × 3 for Patient 2 and arr 5q35.2q35.3(175,822,794–176,904,798) × 3 for Patient 3 (Fig. 1). In all three cases the 5q duplication was mediated by homologous low copy repeats.
Array-CGH on Patient 1 showed the presence of three other copy number variations, resulted inherited from her mother after QPCR analysis. 5q35.2q35.3 duplication was not present neither in her mother nor in her brother (DNA of the father was not available because he died).
MLPA analysis performed in Patient 2 and her parents showed that the duplication originated de novo. Parents of Patient 3 were also investigated by QPCR showing they presented with a normal copy number, indicating that also in this case the duplication originated de novo.
FISH performed on Patient 1 showed that the duplication was direct (a total of 80 nuclei were analyzed), and excluded a possible insertional translocation of the duplicated region in her relatives (Fig. 3). However, we could not exclude that such balanced rearrangement was present in the father.

DISCUSSION
5q35 duplication encompassing NSD1 has been described in 27 patients (Fig. 1) [Chen et al., 2006; Kirchhoff et al., 2007; Franco et al., 2010; Zhang et al., 2011; Dikow et al., 2013; Rosenfeld et al., 2013; Zilina et al., 2013]. Twelve of them have been recently published in Rosenfeld et al. [2013] and four were familiar cases [Rosenfeld et al., 2013]. Nine have been more recently published in Dikow et al. [2013] including six familiar cases [Dikow et al., 2013]. All but two (Subject 8 in Rosenfeld et al. [2013] and Patient 7 in Dikow et al. [2013]) have a whole NSD1 duplication.
Here we report on three other patients with 5q35.2q35.3 duplication of different size to better define the phenotype associated with this rearrangement. A summary of the clinical features of all cases already published is reported in Table I, together with the most relevant clinical characteristics of the three new patients described herein.
| Published cases (27 in total) | Our case 1 | Our case 2 | Our case 3 | 5q35 duplication syndrome (30 pts in total) | |
|---|---|---|---|---|---|
| Size of rearrangement (Mb) | From ∼0.26 to ∼6.4 Mb | 2.1 | 2.08 | 1.08 | From ∼0.26 to ∼6.4 Mb |
| Origin of duplication | 10 n.a. or unknown, 5 de novo, 6 maternal inherited, 2 likely maternal, 1 certainly not maternal | n.d. | De novo | De novo | 11 n.a. or unknown, 7 de novo, 6 maternal inherited, 2 likely maternal, 1 certainly not maternal |
| Sex | 10 F, 14 M | F | F | M | 12 F, 15 M |
| At birth | |||||
| Birth weight | Low | n.a. | 2,900 g | 2,700 g | Low |
| Birth height | Short | n.a. | 50 cm | 48 cm | Short |
| Post-natal features | |||||
| Microcephaly | 22/27 | + | − | + | 24/30 |
| Motor delay | 17/27 | − | + | + | 19/30 |
| Growth retardation | 27/27 | 0 | + | + | 30/30 |
| Intellectual/learning disability | 19/26 (in 1 case feature n.s.) | + | + | + (mild) | 22/29 |
| Seizure | 2/24 (in 3 cases feature n.a.) | − | − | − | 2/24 |
| Congenital heart defects | 2/23 (in 4 cases feature n.a.) | n.a. | n.a. | − | 2/23 |
| Delayed bone age | 5/12 (in 15 cases feature n.a.) | n.a. | n.a. | + | 6/13 |
| Epicanthal folds | 7/27 | − | − | − | 7/30 |
| Hypertelorism | 3/27 | − | + | − | 4/30 |
| Strabismus | 5/27 | − | − | − | 5/30 |
| Thin upper lip | 10/27 | + | + | − | 12/30 |
| Small or down turned mouth | 1/27 | − | + | − | 2/30 |
| Micrognathia | 5/27 | − | − | − | 5/30 |
| Pinnalanomaly | 2/27 | − | − | − | 2/30 |
| Brachydactyly | 4/27 | + (fingers) | − | − | 5/30 |
- F, female; M, male; n.a., not assessed; n.s., not specified; n.d., not determined.
Short stature and growth retardation were present in all patients, conversely to Sotos syndrome characterized by pre and postnatal generalized overgrowth. Microcephaly occurred in 24 out of 30 patients (80%), in contrast with the presence of macrocephaly in Sotos syndrome from mild to severe intellectual disability was present in 22 out of 30 patients (73%) and motor delay in 19 out of 30 (63%). Delayed bone age, in opposite to advanced bone age of Sotos syndrome, was present in 6 out of 12 (50%) subjects in whom bone age was determined, indicating that this feature is relatively common among patients affected by 5q35 duplication syndrome.
A facial phenotype is not immediately recognized, but accurate phenotype analysis may reveal subtle dysmorphisms that are opposite to those observed in Sotos patients. For instance, micrognathia instead of prognathism was present in 5 out of 30 patients (17%); moreover, about 40% of duplicated patients (12 out of 30) presented with thin upper lip vermilion, the most frequent dysmorphic feature (see Table I). Epicanthal folds were also present in 7 out 30 patients.
Brachydactyly was present in five out of 30 cases. Interestingly, our Patients 1 and 3 had small hands and in particular Patient 3 had a metacarpophalangeal profile specular to that observed in Sotos syndrome where large hand and a disharmonic maturation of phalanges and carpal bones have been reported [Butler et al., 1988]. Important major depressive syndrome, starting when she was 25 years old, was reported in our Patient 1 and also in the mother of Subject 4 in Rosenfeld et al. [2013]. It is interesting to note that severe psychotic symptoms was reported in an adult subject with Sotos syndrome [Kessler and Kraft, 2008] and anxiety has been reported in patients with Sotos syndrome-2 [Malan et al., 2010].
The duplication breakpoints of our patients fell between Sos-PREP and Sos-DREP subunits A in Patients 1 and 2 and between Sos-PREP and Sos-DREP subunits F in Patient 3, strongly suggesting NAHR as the underlying mechanism of these genomic imbalances. Few familial (parent offspring) cases have been reported for Sotos syndrome so far, while at least 10 families in which a 5q35 duplication segregates with the disease have already been described [Dikow et al., 2013; Rosenfeld et al., 2013].
In conclusion, 5q35.2q35.3 duplication is associated with a phenotype characterized by short stature, developmental delay and microcephaly without typical facial or osseous anomalies. Some clinical features of this novel syndrome appear to be reciprocal to those observed in Sotos syndrome, indeed suggesting that NSD1 is the dosage-sensitive gene responsible for both these conditions. Some authors argued that considering the 5q35 microduplication syndrome as “reversed Sotos phenotype” is inappropriate since many other chromosomal imbalances have been reported in association with microcephaly or short stature and no “reversed facial features” are present in duplicated patients [Dikow et al., 2013]. Finally, follow-up studies and further data about new patients with a 5q35.2q35.3 duplication are needed to clarify if depression in adult life is also a common symptom.