

Role of epicardial adipose tissue in heart failure with preserved ejection fraction: An emerging molecular mechanism and therapeutic potential
Zhongwen Qi and Dan Wu contributed equally to this work
SOURCES OF FUNDING: This work is supported by the National Natural Science Foundation of China (No. 82405358) and the China Postdoctoral Science Foundation (No. 2023M733915) and Evidence-based research project at Xiyuan Hospital, China Academy of Chinese Medical Sciences (No. XYZX0204-04)
Summary
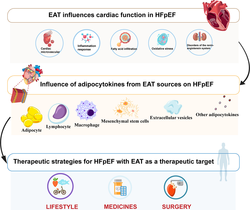
With the evolving landscape of diseases, heart failure with preserved ejection fraction (HFpEF) now encompasses more than half of all heart failure patients. This condition is clinically diverse, involving multiple organ systems and often occurring alongside the aging process. To deeply investigate the common pathogenesis of HFpEF and to explore new therapeutic approaches is of great significance for the treatment of HFpEF. Epicardial adipose tissue (EAT) is not only a dynamic organ with biological functions but also physically adjacent to the myocardium and coronary arteries, endowing it with unique properties as a visceral fat depot. During pathology, EAT can secrete adipocytokines via paracrine mechanisms, establishing direct communication with the heart and vascular, thereby impacting cardiac function. This review aims to elucidate the intricate relationship between EAT and cardiac function in HFpEF, delineate the roles of adipocytes, macrophages, lymphocytes, and stem cells within EAT in HFpEF, and summarize the progress in research regarding drug therapies targeting EAT for HFpEF treatment.
Abbreviations
-
- ACE2
-
- angiotensin (Ang) converting enzyme 2
-
- ADSC
-
- adipose mesenchymal stem cells
-
- ANT2
-
- adenine nucleotide translocase2
-
- BAT
-
- Brown adipose tissue
-
- BMI
-
- body mass index
-
- baPWV
-
- Brachial-ankle pulse-wave velocity
-
- CAMP
-
- cyclic adenosine monophosphate
-
- CMR
-
- cardiac magnetic resonance
-
- EAT
-
- Epicardial adipose tissue
-
- ECM
-
- extracellular matrix
-
- FABP4
-
- fatty acid transporters such as fatty acid-binding-protein 4
-
- FFAs
-
- free fatty acids
-
- GM-CSF
-
- granulocyte-macrophage colony-stimulating factor
-
- GLP-1 Ras
-
- Glucagon-like peptide-1 receptor agonists
-
- HAECs
-
- human aortic endothelial cells
-
- HIF-1α
-
- hypoxia-inducible factor 1-α
-
- HFrEF
-
- heart failure with reduced ejection fraction
-
- HFpEF
-
- heart failure with preserved ejection fraction
-
- HGF
-
- hepatocyte growth factor
-
- IFN-γ
-
- interferon-γ
-
- MCP-1
-
- monocyte chemotactic protein-1
-
- PDGF-BB
-
- platelet-derived growth factor BB
-
- PGC-1α
-
- peroxisome proliferator-activated receptor γ (PPAR-γ) coactivator 1-α
-
- PCSK9
-
- proprotein convertase subtilisin/kexin type 9
-
- STAT3
-
- signal transducer and activator of transcription 3
-
- MAO
-
- trimethylamine N-oxide
-
- TGF-β
-
- transforming growth factor-β
-
- TLR
-
- Toll-like receptor
1 INTRODUCTION
For decades, HFpEF proved an elusive entity to treat1, 2. Consequently, drugs proven effective in improving the prognosis of heart failure with reduced ejection fraction (HFrEF) have not shown significant benefits in HFpEF3. Obesity plays an important role in patients with HFpEF. Although body mass index (BMI) identifies the presence of obesity, this index provides limited information on visceral fat content, which may be more relevant to the pathophysiology of heart failure4, 5. Recent data suggest that accumulation of EAT is associated with the onset, symptoms, and prognosis of HFpEF6. EAT in patients with obesity can trigger systemic inflammation, leading to microcirculatory dysfunction and subsequent myocardial fibrosis and cardiac remodeling7, 8. Structurally, energy transmission between the epicardium and myocardium relies on shared microcirculatory structures9, 10. Consequently, systemic chronic inflammation or metabolic disorders accelerating EAT accumulation and the release of inflammatory cytokines and adipocytokines may constitute key pathogenic mechanisms underlying HFpEF, contributing to microvascular dysfunction and myocardial fibrosis11, 12.
The accumulation of EAT has consistently been associated with left ventricular hypertrophy, diastolic dysfunction, and dilatation of the atria, which are considered typical hallmarks of HFpEF. In addition, EAT has been related to increased cardiac filling pressures and exercise intolerance in HFpEF13, 14. Additionally, EAT as an active regulator of energy metabolism, storing excess energy as triglycerides within lipid droplets and releasing it into the circulation as free fatty acids (FFAs). In the context of overaccumulation, EAT disrupts myocardial metabolism, leading to detrimental lipotoxicity and proinflammatory responses10, 15. Hence, we describe the physiology of EAT and the potential mechanisms of EAT in HFpEF. A series of summaries is also provided on how EAT may be involved in the complex mechanisms of HFpEF. Finally, we outline potential therapeutic strategies, knowledge gaps, and directions for further research.
2 HOW EAT RELATES TO HFPEF AND ITS POTENTIALLY SIGNIFICANT ROLE IN THE PATHOPHYSIOLOGY OF HFPEF
2.1 Role of EAT on HFpEF pathophysiology
EAT exhibits a dual composition of brown adipose tissue (BAT) and white adipose tissue (WAT)16. Most of the fat in the EAT is WAT, and BAT makes up a small portion of the EAT, which is characterized by an abundance of mitochondria that produce large amounts of heat. In addition, the volume of BAT is negatively correlated with cardiovascular events, and the opposite is true for WAT17. A recent observational study showed that certain key proteins associated with adipose tissue browning are highly expressed in the EAT. However, BAT volume decreases with increasing obesity, which is very common in HFpEF18. BAT-characterized EAT plays a crucial role in maintaining free fatty acids (FFAs) homeostasis by controlling fatty acid transporters such as fatty acid-binding-protein 4 (FABP4)19. Moreover, it can augment the mitochondrial uncoupling protein-1 (UCP-1) metabolic activity by uncoupling adenosine triphosphate (ATP) synthesis. This activation is facilitated through peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α (PGC-1α), offering protection against hypothermia to the heart and coronary arteries under conditions of chronic cold exposure20, 21. BAT-characterized EAT correlates with elevated plasma levels of trimethylamine N-oxide (TMAO), which significantly reduces levels of phosphocreatine and ATP in cardiac tissue. This effect arises from TMAO's inhibition of mitochondrial complex IV, consequently contributing the development of heart failure22. Increased EAT volume may negatively affect the left ventricle (LV), compromising its distensibility and contributing to the onset of HFpEF23, 24. When the local microenvironment is modified, numerous deleterious adipokines (including pro-inflammatory and pro-fibrotic) are produced, which may diffuse ultimately to various levels of the vasculature via endocrine or paracrine pathways25, 26 This process contributes to myocardial lipotoxicity and subsequent cardiac dysfunction. Increasing evidence indicates a substantial enlargement of EAT in patients with HFpEF compared to the healthy population and patients with HFrEF. Notably, in patients with obesity with HFpEF, EAT volume is notably elevated, manifesting more severe pulmonary hypertension and increased cardiac filling pressures14, 27. Moreover, heightened EAT is associated with worse hemodynamic impairment, elevated levels of proinflammatory markers, and endothelial dysfunction28, 29. Furthermore, studies have shown that EAT is strongly associated with new-onset HFpEF and increased mortality30, 31. Importantly, this association remains significant after adjusting for factors such as body mass index (BMI), severity of heart failure, and comorbidities32, indicating a direct involvement of EAT in the pathophysiology of HFpEF. An interesting finding revealed that aged females demonstrated higher EAT, and the parameter of left ventricular diastolic insufficiency was significantly associated with increased EAT thickness. This correlation presents compelling evidence for a heightened prevalence of HFpEF among older women27, 33. Moreover, brachial-ankle pulse-wave velocity (baPWV) was higher in HFpEF patients with EAT ≥3.55 mm, suggesting a potential independent association between EAT and arterial stiffness in HFpEF34. EAT has shown other diagnostic and prognostic properties, in addition to its role in mediating inflammation, cardiovascular dysfunction, and fibrosis in the HFpEF population.
2.2 Quantification and characterization of epicardial adipose tissue for HFpEF assessment
Noninvasive visualization of EAT is accomplished in most conditions by CMR, CT, or transthoracic echocardiography. However, the application of magnetic resonance imaging for evaluating adipose tissue, including EAT thickness and the total EAT volume, shows a favorable correlation35. A study conducted by Chinese scholars, comprising 147 patients with HFpEF who underwent standard cardiac magnetic resonance (CMR) examination. Multifactorial logistic regression analysis further revealed a stronger correlation between the EAT of the Right Ventricular Free Wall (RVFW) and the total EAT volume compared to the EAT of the atrioventricular groove. Therefore, the EAT of RVFW may serve as a relevant indicator for the assessment of HFpEF36, potentially linked to EAT-induced myocardial fibrosis and coronary microvascular obstruction, resulting in impaired left ventricular diastolic function37. Another study of HFpEF in the elderly revealed a correlation between different compartments of adipose tissue and the LV eccentricity index. Specifically, only the patients with an increased EAT had a direct relationship with the LV eccentricity index, while abdominal fat did not influence this result38. In another retrospective study involving 379 patients with coronary artery disease undergoing hemodialysis without heart failure at the baseline, 142 patients (37.5%) progressed to HFpEF during a mean follow-up of 4.3 years. At the time of HFpEF development, EAT thickness was elevated compared to patients who did not develop heart failure. Subsequently, during an extended observation period, a linear relationship was observed between EAT increase and HFpEF development over time39. The same results were reported in another cross-sectional study where EAT emerged as an independent predictor of HFpEF, irrespective of left atrial volume index, left ventricular weight index, waist circumference, and E/é40. In the Framingham study involving 1554 patients, EAT thickness measured in the RVFW by CMR showed a correlation with new-onset HFpEF. Furthermore, it was observed that NT-proBNP may partially mediate the effect of thickened EAT on the development of heart failure41.
In an international cohort study, the investigators utilized Doppler echocardiography to classify 182 patients with HFpEF into two groups based on EAT thickness: EAT ≥9 mm and EAT <9 mm. Patients with increased EAT demonstrated higher BMI, smaller left ventricular end-diastolic volume (LVEDV), smaller left atrial (LA) volume, and a trend toward lower Kansas City Cardiomyopathy Questionnaire (KCCQ) scores28. These findings suggest that the functional analyses of the myocardium can be performed in addition to EAT quantification42.
3 POTENTIAL MECHANISMS BY WHICH EAT INFLUENCES CARDIAC FUNCTION IN HFPEF
3.1 Inflammation response
Excessive accumulation of EAT represents an inflammatory state primarily influenced by EAT-derived macrophages, which release pro-inflammatory factors impacting cardiac function43. Previous studies have shown that M1 macrophages increase and M2 macrophages decrease within EAT of patients with obesity, resulting in an elevated M1/M2 ratio. This imbalance facilitates the translocation of NF-κB to the nucleus via macrophage Toll-like receptor (TLR) 2/4, causing a significant upregulation of pro-inflammatory factors, such as IL-1β, TNF-α, and monocyte chemotactic protein-1 (MCP-1). These factors contribute to cardiac cardiological remodeling by acting on the cardiac microvasculature and myocardium through autocrine and paracrine mechanisms44, 45. In diabetes mellitus patients, there is an aggregation of activin A, angiopoietin-2, and CD14-positive monocytes within EAT. This aggregation triggers SMAD2 phosphorylation, a downstream target of activin A signaling, and also influences Akt-Ser473 phosphorylation. Consequently, myocardial fibrosis and cardiac contractile function are affected46. In addition, obesity induces the formation of NLRP3 inflammasomes, fostering an inflammatory environment within the EAT of patients with obesity and heart failure. This inflammatory environment promotes the release of inflammatory factors, ultimately contributing to cardiac remodeling47, 48.
3.2 Oxidative stress
When myocardial energy is adequate, EAT stores fatty acids. However, in cases of FFAs overload (in patients with obesity), there is an overactivation of β-oxidation. This leads to the production of reactive oxygen species (ROS) and alterations in the regulation of sarcoplasmic reticulum calcium ATPase, contributing to diastolic dysfunction49. Additionally, the excessive ROS can also trigger the NF-κB inflammatory pathway in an early phase50. Uncoupling protein 1 (UCP-1) in EAT serves to mitigate the oxidative stress imbalance by promoting thermogenesis, thereby assisting in maintaining tissue homeostasis. The diminished expression level of UCP-1 in pathological states may represent an important mechanism underlying the development of oxidative stress imbalance in EAT and its consequent impact on myocardial injury51.
3.3 Cardiac microvascular dysfunction
Cardiac microvascular dysfunction plays a pivotal role in the pathogenesis of HFpEF. Systemic chronic inflammation is a critical factor triggering disturbances in the microcirculation. Moreover, the pathological accumulation of EAT exacerbates microvascular dysfunction and promotes myocardial fibrosis52. Increased thickness of EAT is not only associated with left ventricular hypertrophy and diastolic dysfunction but also with microvascular endothelial dysfunction28. In individuals with diabetes and HFpEF, EAT contributes to cardiac microvascular inflammation, endothelial dysfunction, and alterations in cardiac and vascular repair mechanisms. Additionally, oxidative stress impacts the availability of nitric oxide (NO), cyclic guanosine monophosphate (cGMP) content, and protein kinase G (PKG) activity in adjacent cardiomyocytes (NO-cGMP-PKG)53, 54. Microvascular endothelial dysfunction leads to reduced NO bioavailability and myocardial PKG content, affecting neighboring cardiomyocytes and cardiac fibroblasts via the sGC-cGMP-PKG signaling pathway55, 56. These alterations ultimately drive changes in cardiac structure and function, leading to the development of HFpEF57. Dapagliflozin treatment has been shown to mitigate hypertension and reverse LV remodeling in HFpEF model pigs, partly by dampening sympathetic tone in the aorta. This effect inhibits the release of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), as well as the activation of the NO-cGMP-PKG pathway58. Low-activity protein kinase G mediates myocardial hypertrophy and increases the occurrence of preserved heart failure, primarily due to hypophosphorylation of actin and the accumulation of collagenous extracellular matrix (ECM).
3.4 Fatty acid infiltration
EAT exhibits heightened activity in the production, absorption, and degradation of FFAs compared to other fat depots59, 60. The heart relies heavily on β oxidation of long-chain fatty acids for energy to maintain contractile function, the excessive accumulation of FFAs in EAT surpasses the myocardium's ability to oxidize FFAs, resulting in FFAs entering cardiomyocytes, a phenomenon often termed cardiac lipotoxicity. This lipid overload within the expanded EAT leads to cardiac dysfunction35, 61, 62, characterized by heightened excessive oxidative stress, endoplasmic reticulum stress, mitochondrial dysfunction, and ultimately to left ventricular diastolic dysfunction63. Palmitic acid is an FFA secreted by EAT. In experimental models using extracted neonatal rat cardiomyocytes, intervention with palmitic acid demonstrated a decreased ability for fatty acid oxidation alongside increased intracellular triglycerides (TG) and ceramides accumulation within cardiomyocytes. These lipid alterations are linked with apoptosis, suggesting that FFAs can induce cardiomyocyte apoptosis and subsequent decline in cardiac function64. Moreover, palmitic acid exerts deleterious effects on cardiomyocytes by increasing oxygen consumption and inducing hypoxia-inducible factor 1-α (HIF-1α) expression in adipocytes via increased activity of adenine nucleotide translocase (ANT2), a mitochondrial membrane uncoupling protein65. The excessive FFAs released by accumulated EAT not only trigger macrophage infiltration but also prompt the secretion of pro-inflammatory adipocytokines such as leptin, TNF-α, IL-6, IL-1β, and resistin. This inflammatory milieu is further compounded by a concurrent decrease in adiponectin66.
3.5 Disorders of the renin-angiotensin system
The renin-angiotensin system (RAS) is a crucial mechanism for cardiac remodeling, with angiotensin (Ang) converting enzyme 2 (ACE2) serving as a critical regulator by metabolizing Ang II to Ang 1–7. To explore the impact of ACE2 deficiency on cardiac health, researchers subjected ACE2-deficient (ACE2KO) mice to a high-fat diet for 6 months. Their findings revealed increased inflammation within EAT, characterized by a macrophage proinflammatory phenotype. This inflammatory milieu was concurrent with a reduction in cardiac adiponectin and an escalation in cardiac lipotoxicity, ultimately culminating in worsened cardiac diastolic function, a condition more predisposed to HFpEF44 (Figure 1).

4 POTENTIAL INFLUENCE OF EAT-DERIVED CYTOKINES ON HFPEF
4.1 Adipocytokines of adipocyte source in EAT
Adiponectin, an endocrine factor of adipocyte origin, plays a crucial role in regulating the local inflammatory microenvironment within the myocardium. In a co-culture setting involving adipocytes and human aortic endothelial cells (HAECs), TNF-α induced injury to HAECs was investigated. It was found that adiponectin had a more stable binding capacity toward HAECs compared to adipocytes. Furthermore, adiponectin exerted inhibitory effects on IKA-α phosphorylation and subsequent NF-kB activation within HAECs. Notably, adiponectin did not affect the phosphorylation status of Jun-n-terminal kinase, p38 kinase, and Akt kinase. Additionally, concurrent with adiponectin action, there was an accumulation of cAMP, an effect modulated by protein kinase A (PKA) inhibitors67. These findings suggest that adiponectin, secreted by adipocytes, acts as a stabilizer of NF-κB, thereby regulating the local inflammatory response in endothelial cells and potentially influencing the progression of HFpEF via the interaction between cAMP-PKA and NF-kB signaling pathways. Recognized as a cardioprotective factor, adiponectin is implicated in ameliorating patients in heart failure by engaging with the adiponectin receptors AdipoR1 and AdipoR2. This interaction facilitates enhanced insulin-stimulated Akt phosphorylation and initiates activation of the PPAR-α/AMPK pathway, leading to increased glucose uptake and fatty acid oxidation68. Consequently, interventions involving recombinant adiponectin proteins hold promise for cardiovascular disease.
Leptin, primarily secreted by white adipocytes, exerts its influence by binding to receptors in the hypothalamus, where it regulates appetite and improves peripheral insulin resistance69, 70. Beyond its metabolic roles, leptin also functions as an inflammatory cytokine. Specifically, individuals with coronary artery disease carrying the leptin gene (type AA) and the leptin receptor gene (type RR) were found to have a three folds higher risk of HFpEF compared to the general population71. In addition, leptin exerts direct effects on cardiomyocytes, impacting intracellular calcium storage and consequently impairing myocardial relaxation. Moreover, it stimulates collagen synthesis by enhancing aldosterone secretion, leading to myocardial fibrosis72. In an intriguing in vivo modeling study, leptin was shown to modulate gene expression patterns, promoting the down-regulation of pro-inflammatory genes and up-regulation of antioxidant genes. This resulted in decreased levels of reactive oxygen species (ROS) and consequent reduction in myocardial damage73.
4.2 Adipocytokines of macrophage source in EAT
miRNAs originating from EAT play a pivotal role in crosstalk between EAT and distant target tissues via blood circulation74. Macrophages in EAT secrete a variety of miRNAs, among which miR-21 stands out for its contribution to myocardial fibrosis. This occurs through the facilitation of endothelial-mesenchymal transition mediated by the action of transforming growth factor-β (TGF-β)75. Conversely, miR-17-5p secreted from EAT targets recombinant transforming growth factor beta receptor II (TGFBR2) and signal transducer and activator of transcription 3 (STAT3). By inhibiting adipocyte proliferation, miR-17-5p indirectly enhances cardiac function and serves as a biomarker for heart failure76. Furthermore, the expression of miR-27a, a lipid-forming inhibitor via peroxisome proliferator-activated receptor gamma (PPAR-γ), can be upregulated by TNF-α through the NF-κB pathway77. This pro-inflammatory state induced by miR-27a upregulation contributes to the progression of heart failure. Obesity, often associated with hypoxia, leads to adipose tissue dysfunction, insulin resistance, and inflammation65. During tissue hypoxia, HIF-1α is activated. HIF-1α promotes the recruitment of M1 macrophages, which heightens metabolic demands and fosters an inflammatory environment within adipose tissue78, and hypoxic HIF-1α exacerbates myocardial fibrosis and diastolic dysfunction79. Macrophages in EAT secrete CD5L, a factor inhibiting macrophage apoptosis, which may activate the TLR4/NF-κB pathway in the adjacent cells. This activation results in the production of pro-inflammatory cytokines, further perpetuating the inflammatory cascade implicated in cardiovascular pathology80, 81.
4.3 Adipocytokines of lymphocyte source in EAT
In patients with heart failure, EAT undergoes significant immune activation, mainly characterized by the aggregation of T-lymphocytes. Indeed, the immune cells in EAT of patients with heart failure exhibit a notable aggregation, with a predominant presence of interferon-γ (IFN-γ) positive effector memory T lymphocytes82. This immune response is driven by adipose-retained M1 macrophages, which promote the activation of effector Th1 cells, including CD8 + cytotoxic T cells. These activated T cells play a crucial role in producing IFN-γ and stimulating the synthesis and secretion of TNF-α, thereby activating the Janus Kinase (JAK) signaling and the activator of STAT3 signaling pathway83. Increased infiltration of adipose tissue by T cells and macrophages also affects adipocyte function by modulating the secretion of adipokine84. Furthermore, the immune response involves the release of various cytokines by Th1 and Th2 cells, such as IFN-γ and interleukin-4 (IL-4), which influence the differentiation of T cell subsets. In addition, Th17 cells are strong mediators of local tissue inflammation, secreting cytokines like IL-17, IL-21, IL-22, and IL-2385. However, the activation of Th17 cells can lead to persistent adipose tissue inflammation through cytokine release86. Early CD8 cells are also implicated in this immune response, as they can produce granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine of activating M1 macrophages. This activation, in turn, fosters a pro-inflammatory microenvironment within EAT, contributing to the perpetuation of the inflammatory cascade associated with heart failure87.
4.4 Mesenchymal stem cells of EAT
Mesenchymal stem cells (MSCs) in EAT possess the capability to migrate toward the myocardium, where they can undergo differentiation into fibroblasts88. This process contributes to a decrease in LV compliance, as evidenced by impaired LV relaxation or increased stiffness. Furthermore, the protective effect of MSC-derived extracellular vesicles (EVs) on cardiac remodeling has been reported in a mouse model of pressure overload. Enhanced release of EVs has been associated with improved ejection fraction, short-axis shortening rate, and cardiac weight, particularly when MSCs are administered independently89. Moreover, the injection of adipose-derived mesenchymal stem cells (ADSC) overexpressing N-cadherin into mice with heart failure was found to increase left ventricular ejection fraction (LVEF) and reduce fibrosis. In vitro experiments have elucidated that the overexpression of N-cadherin facilitates ADSC-cardiomyocyte adhesion and migration, augments angiogenesis and cardiomyocyte proliferation, and mediates ADSC migration and paracrine angiogenesis. These effects are achieved through the modulation of up-regulated expression of matrix metallopeptidase-10/13 (MMP-10/13) and hepatocyte growth factor (HGF)90.
4.5 Extracellular vesicles in EAT
EAT serves as a significant source of bioactive factors that can influence adjacent cardiomyocytes and vascular91, 92. In the context of heart failure, several studies have elucidated the role of microRNAs (miRNAs) carried by Evs derived from adipocytes and adipose tissue macrophages in contributing to cardiac fibrosis, dysfunction, hypertrophy, and ultimately heart failure. EV-miR-23a-3p, secreted by adipocytes isolated from Ang II-induced heart failure mice, has been implicated in cardiac fibrosis and dysfunction. Mechanistically, miR-23a-3p has been shown to accelerate excessive collagen deposition by targeting Ras-proximate-1 (RAP1), a GTPase involved in the transformation of cardiac fibroblasts into myofibroblasts and collagen deposition93. Adipose tissue macrophage-derived exosome-miR-29a is translocated into cardiomyocytes, leading to myocardial hypertrophy and eventually heart failure by activating the AKT/mTOR pathway94. Additionally, it inhibits autophagy in cardiomyocytes through the PTEN/AKT/mTOR pathway, impairing the removal of damaged cells and potentially perpetuating the inflammatory process95. However, there are also reports supporting the cardioprotective role of miR-29a in reducing cardiac hypertrophy by regulating PPAR-γ96. Another study demonstrated that miR-29a knockout mice has been associated with the development of multiple cardiac pathological features. This phenotype is mediated through the miR-29a/PGC-1α regulatory circuit, which affects cardiac mitochondrial metabolism. Dysregulation of this circuit interferes with myocardial fibrosis and hypertension, contributing to HFpEF97.
4.6 Other adipocytokines in EAT
Adrenomedullin (AM), an adipokine secreted by EAT, is a vasoactive polypeptide regulated by cyclic adenosine monophosphate (CAMP), nitric oxide (NO), the renal prostaglandin system, and vascular endothelial growth factor98. AM diffuses into adjacent myocardium or vasculature, where it interacts with endothelial cells and vascular smooth muscle cells (VSMCs)25. Upon infiltration into the vascular endothelium, AM performs integrative functions, reducing vascular permeability by stimulating NO synthesis and counteracting the effects of angiotensin II and endothelin I99. In addition, AM inhibits the expression of matrix metalloproteinase-2 (MMP-2), leading to extracellular matrix degradation, suppression of excessive collagen fiber synthesis,100 and improvement in myocardial fibrosis and ventricular remodeling.
Taken as representatives of thermogenic genes, UCP1, PGC-1α, and PR-domain missing 16 (PRDM16) are believed to serve as specific markers of brown fat, governing oxidative metabolism and mitochondrial biogenesis, and contributing significantly to cardioprotection. The expression of thermogenic factors in EAT is diminished in patients with HFpEF in contrast to those with HFrEF. This variation exposes the myocardium to an excessively lipotoxic environment and promotes proinflammatory pathways101.
Omentin-1, an adipokine expressed predominantly by EAT, improves insulin sensitivity and exhibits anti-inflammatory and cardiovascular protective properties 102. As an antifibrotic adipokine, Omentin-1 mitigates myocardial fibrosis in heart failure by attenuating the TGF-β signaling pathway103. In a high-fat diet-fed mouse model, treatment with omentin-1 suppressed the Akt pathway, resulting in decreased levels of pro-inflammatory cytokines such as NF-kB and a reversal in macrophage polarization, thereby conferring a cardioprotective effect104. Omentin-1 inhibited angiotensin II-induced monocyte/macrophage migration and platelet-derived growth factor BB (PDGF-BB) induced proliferation of VSMCs105.
A recently identified cardioprotective adipokine, vaspin, which protects the myocardium by impeding the PI3K/AKT/mTOR pathway and diminishing TNF-α induced apoptosis106. Furthermore, it mitigates pathological myocardial hypertrophy by suppressing the PI3K/AKT/mTOR pathway to modulate cardiac senescence107. In addition, vaspin inhibits NLRP3 inflammasome activation through the induction of autophagy, thereby sustaining the suppression of inflammatory factor release108 (Figure 2).

5 POTENTIAL TREATMENT OPTIONS FOR HFPEF TARGETING EAT IN ITS EARLY STAGES
5.1 Statins
Although statins are widely recognized for their anti-inflammatory effects in patients with atherosclerosis, recent findings suggest their potential to ameliorate the inflammatory process within EAT, thereby exerting favorable effects on cardiac remodeling in a mouse model of HFpEF109 and improving cardiac diastolic dysfunction in clinical patients110. Notably, in two large randomized controlled clinical trials involving patients with dyslipidemia, statins reduced the risk of new-onset heart failure111, although no significant benefit on morbidity and mortality was observed in patients with HFrEF112, 113. In a cohort of 9140 patients with HFpEF, those treated with statins exhibited a higher one-year survival rate compared to untreated patients, suggesting a potential mortality-reducing effect in HFpEF114. In addition, echocardiography-derived measurements of EAT thickness revealed that statin-treated patients displayed reduced EAT thickness and inflammatory markers secretion compared to untreated counterparts115. Moreover, as part of the EAT secretome, recombinant proprotein convertase subtilisin/kexin type 9 (PCSK9) is involved in EAT-induced inflammation116. Thus, PCSK9 inhibitors may mitigate abnormal EAT expansion. A non-randomized cohort study reported a 20.39% reduction in EAT thickness following 6 months of PCSK9 inhibitor (evolocumab or alirocumab) administration in patients117. In hyperlipidemic post-menopausal women, intensive statin therapy (atorvastatin 80 mg/d) was associated with accelerated regression of EAT thickness, an effect seemingly independent of LDL lowering and possibly attributed to the result of other actions of statins (anti-inflammatory effects)118. Additionally, in a retrospective cohort study enrolling patients treated with successful percutaneous coronary intervention (PCI), consecutive treatment with atorvastatin (20 mg/d) was more effective in reducing EAT thickness compared to the simvastatin/ezetimibe group119.
5.2 Metformin
In addition to the hypoglycemic effect of metformin, in recent years the drug has been suggested for the treatment of systemic inflammatory diseases. This expanded utility is mainly attributed to its ability to activate adenosine monophosphate-activated protein kinase (AMPK), thereby inhibiting the pro-inflammatory effects of nuclear factor-kappa B (NF-κB)120. Additionally, metformin has been found to stimulate the release of adiponectin, an adipokine with anti-inflammatory properties 121. Adiponectin, in turn, acts to suppress the secretion of inflammatory cytokines from EAT. In an animal model of diabetic cardiomyopathy, metformin was observed to delay myocardial electrical remodeling and structural remodeling. These effects are thought to arise from the attenuation of cardiomyocyte hypertrophy, reduction of myocardial fibrosis, and protection against microvascular injury122-124. In clinical observations of patients with established heart failure, metformin has shown promise in reducing the risk of re-hospitalization for heart failure and decreasing overall mortality rates125-127. While administration of metformin at standard doses (500–1000 mg twice daily) for 3–6 months did not significantly reduce EAT thickness in patients with T2DM128, increasing the therapeutic dose resulted in a mild reduction in EAT thickness129, Despite the lack of significant impact on EAT thickness, studies have suggested that metformin may still confer benefits in terms of reducing mortality and improving left ventricular hypertrophy and diastolic dysfunction in patients with HFpEF130.
5.3 Thiazolidinediones
Thiazolidinediones function as agonists of PPAR-γ, facilitating the differentiation of precursor adipocyte131. Beyond regulating the energy supply of the epicardium and adiponectin secretion, PPAR-γ also alleviates inflammation in adipocytes and cardiomyocytes, cellular hypertrophy, and oxidative stress132. Administration of rosiglitazone to obese Zucker rats resulted in a significant upregulation of PGC-1α, leading to accelerated browning of EAT and increased fat turnover133. One discovery was that thiazolidinediones reduced EAT inflammation while concurrently reducing cardiac inflammatory biomarkers, independent of the drug's glucose-lowering effects134, 135. However, thiazolidinediones are potent stimulators of renal tubular sodium reabsorption, thereby causing circulating sodium retention136. In several randomized clinical trials, sustained use of thiazolidinediones increased the risk of re-hospitalization for heart failure events in patients with HFrEF137-139. In contrast, thiazolidinediones did not increase heart failure events in the Large Scale Clinical Cardiovascular Outcomes Trial involving patients with HFpEF140. This beneficial outcome may be related to the anti-inflammatory properties of thiazolidinediones, which ameliorate EAT inflammation, thereby enhancing diastolic dysfunction. This anti-inflammatory effect counteracts the detrimental effect of sodium retention in the circulation141.
5.4 Glucagon-like peptide-1 receptor agonists
Previous works have documented the presence of GLP-1R expression in EAT. Targeting GLP-1R has shown promise in reducing local adipogenesis, enhancing fat utilization, and promoting brown fat differentiation142. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) have been found to improve cardiac function and attenuate myocardial injury in a mouse model with previous atherosclerosis143. In experimental HFpEF models, GLP-1 RAs mediate adipocyte GLP-1 receptor signaling, inhibit adipocyte proliferation142, reduce inflammation, and support normal energy metabolism. These effects contribute to mitigating myocardial inflammation and myocardial fibrosis144, 145. Short-term treatment with GLP-1 RAs, alongside weight loss, has been observed to induce the redistribution of excess adipose tissue. This redistribution potentially holds implications for improving cardiovascular risk, particularly in patients with type 2 diabetes146, 147. Liraglutide, semaglutide, and dulaglutide have demonstrated rapid reductions in EAT among patients with type 2 diabetes128, 148. A recent preclinical study of semaglutide found that it improved cardiac diastolic function, left ventricular thickness, and exercise tolerance in an aged obesity-associated HFpEF mouse model. Further analyses, including single-cell transcriptomic, have suggested potential mechanisms related to cardiomyocyte cytoskeletal proteins, and endothelial cell function, as well as reductions in oxidative stress and inflammatory status within adipose tissue149. Nevertheless, it's important to note that in two published randomized clinical trials involving patients with HFrEF, liraglutide was associated with adverse effects on sympathetic nerve activity, leading to worsening clinical heart failure symptoms and severe heart failure150, 151. Conversely, the STEP-HFpEF clinical trial demonstrated that the benefits of semaglutide in patients with obesity-related HFpEF were independent of the reductions in BMI. Additionally, both diabetic and non-diabetic patients experienced similar improvements in parameters such as 6 minutes walking distance and exercise tolerance152, 153. Further large-scale clinical trials across different phenotypes of heart failure are necessary to ascertain the potential therapeutic effect of GLP-1 RAs in patients with HFpEF.
5.5 Sodium-dependent glucose transporters-2 inhibitor
SGLT-2 inhibitors are approved by the US FDA in 2021 as innovative glucose-lowering agents for the treatment of patients with heart failure154, 155. The EMPEROR-Preserved trial evidenced a 21% reduction in the risk of the primary endpoint events and a 27% decrease in heart failure rehospitalization rates among patients with HFpEF (LVEF>40%) treated with empagliflozin compared to placebo156. Furthermore, Empagliflozin notably ameliorated interstitial myocardial fibrosis and aortic stiffness in non-diabetic individuals with HFrEF by diminishing EAT volume157. Various animal studies exploring HFpEF have demonstrated the cardioprotective effects of SGLT-2 inhibitors, which mitigate cardiac function in HFpEF by attenuating adipose tissue inflammation and adipocyte hypertrophy158, as well as improving myocardial hypertrophy and myocardial fibrosis159, 160. Clinical observations have highlighted the positive impact of SGLT-2 inhibitors on abnormal diastolic function in patients with heart failure, with benefits linked to reduced EAT mass and inflammation161, along with a diminished risk of heart failure-related rehospitalization162, 163. Thicker EAT was present in patients with obesity-based HFpEF, hemodynamic abnormalities such as elevated right atrial pressure, pulmonary artery pressure, and pulmonary capillary wedge pressure at rest and during exercise are evident14. EAT accumulation is prevalent in patients with T2DM, obesity, and other comorbidities164. Immunohistochemical analyses have revealed the expression of SGLT-2 in EAT, with notably higher levels in men than in women, whereas its expression in subcutaneous fat is minimal. Dagliflozin-treated EAT samples showed enhanced glucose uptake, decreased secretion of pro-inflammatory chemokines, and improved adipocyte differentiation compared to the insulin-treated group165. In an additional study involving empagliflozin, treatment over 24 weeks resulted in a roughly 20% reduction in EAT thickness among patients with diabetes comorbid with obesity (BMI ≥ 27 kg/m2), independent of weight loss129. Among 14 patients with type 2 diabetes mellitus combined and stable coronary artery disease, myocardial flow reserve could be improved by 4 weeks of treatment with empagliflozin, which was found to reduce EAT thickness by 19%, along with a 21.6% reduction in uptake glucose by EAT, consequently mitigating the inflammatory state of the EAT166. SGLT-2 inhibitors improve cardiac metabolic remodeling by modulating metabolism and inflammation within adipose tissue and preventing myocardial fibrosis through the EAT- SGLT-2 pathway167. Treatment with canagliflozin for 6 months reduced EAT thickness (from 9.3 ± 2.5 to 7.3 ± 2.0 mm) in patients with type 2 diabetes mellitus regardless of its glucose-lowering effect168. Moreover, a 12-week ipragliflozin intervention in non-obese type 2 diabetics (BMI < 25 kg/m2) led to an average reduction in EAT thickness of 13cm3 while concurrently increasing high-density lipoprotein cholesterol level169.
5.6 Anti-cytokine drugs
TNF-α and IL-1β, synthesized and released by EAT, are elevated in patients with HFpEF, but not in those with HFrEF170. In animal models of HFpEF, proinflammatory cytokines and other members of the IL-1 family play a direct role in myocardial fibrosis and diastolic dysfunction, contributing to structural abnormalities in HFpEF171, 172. Intervention targeting these cytokines has shown promise in ameliorating these structural abnormalities in HFpEF. In the D-HART trial, the application of the competitive IL-1 receptor antagonist Anakinra for 14 days in patients with HFpEF led to significant improvements in systemic inflammation and aerobic exercise capacity, albeit with an increase in plasma CRP levels173. However, subsequent phase II clinical trials did not confirm positive outcomes in terms of cardiorespiratory function174. In addition, anti-cytokine therapies have been observed to promote aldosterone synthesis, resulting in sodium retention and exacerbating heart failure175. However, a small randomized controlled trial demonstrated that the interleukin 1 antagonist Anakinra reduced systemic inflammation in patients with rheumatoid arthritis while ameliorating the reduction in adverse exercise tolerance associated with HFpEF173. In additional large randomized controlled trials involving patients with systemic inflammation, the IL-1β antagonist canakinumab was found to decrease pro-inflammatory markers and reduce the risk of heart failure hospitalization176 (Table 1).
| Therapeutic drug | Possible mechanisms | Negative incident | References |
|---|---|---|---|
| Statins | Ameliorate the inflammatory process in EAT | - | [102] |
| Ameliorate cardiac diastolic dysfunction | [103] | ||
| Reduce mortality | [107] | ||
| Metformin | Activation of AMP/NF-κB and inhibiting the anti-inflammatory effect | - | [113] |
| Promoting adiponectin release attenuates EAT inflammatory cytokines | [114] | ||
| Delay myocardial electrical remodeling and structural remodeling | [115, 117] | ||
| Reduce mortality and improve left ventricular hypertrophy | [123] | ||
| Thiazolidinediones | Activating PPAR-γ to alleviate adipocyte and cardiomyocyte inflammation | Sodium retention, and increased risk of re-hospitalization for heart failure | [124, 125] |
| Reduce EAT inflammation and cardiac inflammatory biomarkers | [127, 128] | ||
| GLP-1 RAs | Promote brown fat differentiation | Affect sympathetic nerve activity; Worsening of clinical heart failure symptoms | [135] |
| Inhibit adipocyte proliferation | [136] | ||
| Ameliorate myocardial inflammation and myocardial fibrosis | [137, 138] | ||
| Induce redistribution of excess adipose tissue | [139, 140] | ||
| SGLT-2 inhibitors | Reduce the risk of the primary endpoint event | Urinary system infections | [148] |
| Attenuates adipose tissue inflammation and adipocyte hypertrophy, and improves myocardial hypertrophy | [150-152] | ||
| Improve abnormal diastolic function | [153] | ||
| Increased glucose uptake, decreased secretion of pro-inflammatory chemokines | [158] | ||
| Activating the NO-cGMP-PKG pathway | [160] | ||
| Reduce EAT thickness and increase high-density lipoprotein cholesterol | [162] | ||
| Anti-cytokine | Improve systemic inflammation and aerobic exercise capacity | Raise plasma CRP levels | [166] |
| Reduce pro-inflammatory markers | Promote aldosterone synthesis, and exacerbates heart failure | [169] |
5.7 Non-pharmacological treatment strategies
Lifestyle modifications (including low-calorie diets and exercise training) have been shown to decrease food intake. In a cohort of 20 patients with severe obesity with HFpEF, a 32% reduction in EAT thickness was observed, accompanied by alleviation of left ventricular hypertrophy and diastolic dysfunction following 6 months of calorie restriction and moderate exercise177. A meta-analysis focusing solely on randomized controlled trials demonstrated that endurance training was a particularly effective strategy in reducing epicardial fat reserve178, with exercise training significantly decreasing EAT thickness and serum TNF-α levels, while concurrently increasing adiponectin and enhancing left ventricular myocardial compliance179. The value of bariatric surgery in conferring cardiac benefit varies; a study utilizing a biliopancreatic shunt observed rapid reductions in visceral adipose tissue, with notable reductions in EAT and pericardiac fat at 12 months of assessment180. In vitro experiments revealed that exposing mature human adipocytes to low glucose levels for 96 hours reversed the expression of deleterious adipokines associated with metabolic complications181. Pericardiectomy has been shown to mitigate elevated LV filling pressures resulting from EAT accumulation, presenting a potential option for the treatment of patients with HFpEF undergoing severe LV restriction. This approach has now been successfully validated in untrained dog and pig models of diastolic dysfunction182, 183 (Figure 3).

6 CONCLUSION REMARKS
HFpEF presents as a complex and heterogeneous clinical syndrome with increasing prevalence and incidence, yet significant challenges persist in understanding its pathogenesis and devising effective treatment strategies. Underlying systemic chronic inflammatory conditions or metabolic disorders serve as pivotal contributors to the heightened incidence and pathogenicity of HFpEF. Notably, obesity emerges as a prominent risk factor for HFpEF, with EAT exerting a consequential influence on cardiovascular function within this context 14, 184. Various adipocytokines (pro-inflammatory, pro-fibrotic) emanate from EAT, thereby intricately participating in the progression of HFpEF. Hence, targeting EAT emerges not only as an intervention for obese HFpEF but also as a fundamental pathological substrate for diverse HFpEF presentations. The non-invasive quantification of EAT through imaging methodologies holds promise in predicting cardiometabolic risk and serving as a therapeutic target for intervention. Therefore, exploring the pathogenesis of HFpEF through the perspective of EAT metabolic remodeling presents a new promising target. With the publication of the results from several large-scale randomized controlled trials investigating SGLT-2 inhibitors holds the promise of furnishing elevated-level evidence to bolster HFpEF treatment paradigms. In the future, post-marketing reassessment of drugs ameliorating EAT energy metabolic remodeling. All of the pharmacological effects mentioned above are only in the early stages of exploration, and EAT has some correlation with HFpEF, there is no comprehensive direct evidence confirming clinical potential benefits.
7 KNOWLEDGE GAPS AND FUTURE PROSPECTS
To address the potential value of EAT in HFpEF, future investigations should focus on the following areas: (1) development of non-invasive, quantifiable assessment parameters for EAT to serve as early warning of metabolic risk in HFpEF and prognostication of heart failure; (2) elucidation of the mechanism underlying the differentiation between EAT and paracardiac adipose tissue in the context of HFpEF; (3) development of recombinant proteins targeting adipokines within EAT to harness their protective effects, followed by the design of small molecule chemotherapeutic agents targeting these proteins; (4) conducting large-scale clinical studies to characterize the genetic and biomolecular profiles of EAT and its correlation with HFpEF; (5) investigation into methods for improving microenvironment of EAT and identification of new targets that influence its composition and distribution; (6) examination of the mechanisms governing EAT regulation in various HFpEF phenotypes (e.g., hypertension, diabetes mellitus, kidney dysfunction, and metabolic syndrome) and the gender-specific mechanism underlying HFpEF oneset, with the aim of personalizing treatment strategies (Table 2).
| Future prospects | Existing technologies and areas | Outcomes produced |
|---|---|---|
| Non-invasive, quantifiable assessment parameters for EAT | Cardiac Magnetic Resonance Evaluation of EAT Thickness | Warning of metabolic risk in HFpEF |
| Clarifying possible mechanisms of EAT in HFpEF | Epicardial thickness is correlated with HFpEF | EAT as a new therapeutic target for HFpEF |
| Drug development targeting adipocytokines | EAT-derived cytokines have predictive value for HFpEF | Development of small molecule compounds for the treatment of HFpEF |
| Alterations in the microenvironment surrounding EAT | Expression of cytokines secreted by EAT | Effect of organ-organ interactions on HFpEF |
| Clinical value of EAT in different phenotypes of HFpEF | Correlation of EAT in all HFpEF patients | Individualized treatment for HFpEF heterogeneity |
AUTHOR CONTRIBUTIONS
Zhongwen Qi and Dan Wu contributed to the draft writing. Zhipeng Yan, Qing Wang, and Yiming Li contributed to the literature collection and analysis. Junnan Zhao contributed to the picture preparation. Fengqin Xu contributed to the manuscript revision. All the authors approved the submission.
ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation of China (No. 82405358) and the China Postdoctoral Science Foundation (No. 2023 M733915) and Evidence-based research project at Xiyuan Hospital, China Academy of Chinese Medical Sciences (No. XYZX0204-04). Some of the figure materials are from https://www.pinclipart.com. We thank Dr. Biqing Wang of University College London for editing the language of this manuscript. We also thank Wiley Editing Services for revising the language of this manuscript.
DECLARATION OF COMPETING INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.

