

Mechanistic insight, diagnosis, and treatment of ammonia-induced hepatic encephalopathy
Abstract
Hepatic encephalopathy is a neuropsychological syndrome due to biochemical disturbance of brain function in advanced liver disease patients. Diagnosis and treatment of the condition is very demanding and has negative toll on finances with increased healthcare utilization. The pathophysiology is not completely understood; however, there is evidence that ammonia plays an important role in the etiology. Conventional methods of solely relying on blood ammonia level to diagnose hepatic encephalopathy did not help much; likewise, the use of lactulose alone in treating hepatic encephalopathy has also been discouraged. This paper analyzed the current knowledge regarding the mechanism of how ammonia disrupts the normal brain function as well as the use of latest diagnosing tools including those under development to evaluate the neuropsychiatric state of patients and their quality of life. The efficacies of lactulose and rifaximin combination for short-term and long-term treatment in addition to nutritional interventions and other drugs undergoing clinical trials were also reviewed.
Introduction
Great progress has been made in incorporating science and technology into medical practice; however, a lot more remain to be performed if we are to accurately diagnose and treat all diseases. The human race is faced with threats of emerging diseases, while the old ones are not completely understood. Several cases of liver diseases are reported yearly across the world. In the field of hepatology, liver disease and cirrhosis are the seventh leading cause of death among adults between 25 and 64 years of age in the USA alone.1 A report by the World Health Organization in China concluded that an estimated 90 million people (which is almost 7% of the population) are infected with hepatitis B virus. It further stated that most of those infected do not experience symptoms and are therefore not aware of their status; however, a third of them may develop serious, life-threatening illnesses such as cirrhosis and liver cancer.2 Hepatic encephalopathy (HE), which is a frequent complication and one of the most debilitating manifestations of liver disease,3 presents as a complex spectrum of neurological cognitive disturbance and altered level of consciousness.4 It affects sleep patterns, personality traits, and intellect and disturbs daily routines such as driving, cycling, house chores, exercise, and even typing.5 Recent study by Butterworth into the factors triggering HE suggested that high ammonia level, inflammatory cytokines, and manganese deposition in the basal ganglia among others increase the development of HE.6 Further research by Jones et al. revealed that accumulation of false neurotransmitters (octopamine), activation of the γ-aminobutyric acid, and inhibitory neurotransmitter system cause a detrimental effect on brain enzymes.7 Conditions such as portosystemic bypass or shunt can cause undetoxified blood to be transported directly to the brain, altering chemical composition of brain electrolytes.8 However, none of these conditions have been thoroughly understood, leaving ammonia etiology being considered the main factor in development of HE. Patients with preexisting liver disease and subsequent renal failure experience a rise in ammonia levels because of reduced urea and perivenous glutamine synthesis capacity, resulting in less ability to detoxify ammonia in the liver and decreased ammonia excretion function of the kidneys.9 High ammonia concentration in the brain leads to inflammation of brain cells, elevation of cerebral glutamine, and astroglial swelling, causing increased intracranial pressure, cerebral edema, and disturbance of brain functions.10 Ammonia excretion from the body takes place in several ways. (Fig. 1). Urea cycle, which converts ammonia into urea, is the highest elimination route.11 Whereas ammonia is harmful to the body, urea has been found to be latent and water soluble hence not toxic to the body.12 Factors such as genetic errors in metabolism and disturbance of one or more enzymes involved in urea cycle affect the conversion of ammonia to urea.13 Glutamine synthetase found in several tissues of the body also help convert ammonia with glutamate to glutamine. The ability of the body to trigger the clearance of nitrogenous waste is further disturbed as the lining of the gut microbiota is altered in patients with cirrhosis.14 Several cases of HE induced by liver diseases are reported yearly across the world.15 Treatment goal of HE aims at tackling the precipitating factors as well as reducing the level of ammonia concentration in the blood. Confirming HE has always depended on the level of blood ammonia in the body, and the main drug used for treatment over the years has been lactulose. However, the complexity of the condition calls for diverse diagnosing tools and multi-approach treatment options. To find better strategy for the management of HE, new insight in understanding the mechanism of the effect of ammonia on brain cells is essential. Therefore, in this review paper, we analyzed the possible mechanisms by which ammonia disrupts brain function and summarized recently developed tools for diagnosis as well as the recommended drugs, nutritional interventions, and drugs undergoing clinical trials for short-term and long-term treatment of ammonia-induced HE.

 , normal metabolism of ammonia;
, normal metabolism of ammonia; 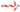 , failure to metabolize ammonia; GABA, γ-aminobutyric acid. [Color figure can be viewed at wileyonlinelibrary.com]
, failure to metabolize ammonia; GABA, γ-aminobutyric acid. [Color figure can be viewed at wileyonlinelibrary.com]The ammonia etiology of hepatic encephalopathy
The ammonia pathogenesis of HE has several explanations. Studies showed that amino acids are not stored in the body but rather synthesized de novo. Catabolism of excess amino acids results in the formation of α-amino groups, which undergoes transamination and subsequent deamination to form ammonia and α-keto acids. Part of the free ammonia is excreted in urine, but chunks of it leave the body via urea cycle.
Ammonia conversion to urea and glutamine
Ammonia is constantly produced in tissues but present in very low levels in the blood because it is converted into a less toxic state by the urea cycle in portal hepatocytes. However, during liver disease, there is decreased capacity for urea synthesis resulting in reduced hepatic clearance of ammonia. Hyperammonemia activates glutamine synthesis from ammonia in extrahepatic tissues. Glutamine synthetase, found in almost all tissues, catalyzes the condensation of ammonia with glutamate to glutamine. Skeletal muscles play an important role as the main alternative organ for ammonia detoxification to glutamine.16 Other organs with recognized glutamine synthetase activities are the brain, the heart, perivenous hepatocytes, and possibly the lungs. Circulatory glutamine released from the brain and skeletal muscle is metabolized largely in enterocytes, immunocytes, periportal hepatocytes, and in the kidneys in the glutaminase reaction to ammonia and glutamate. Glutamine is a major nontoxic interorgan ammonia carrier, but enhanced glutamine presence in the blood tends to activate glutamine catabolism to ammonia in the gut and the kidneys. The kidneys help reduce blood ammonia concentration through ammonia urine excretion, but this process is limited. Severe liver disease overburdens the kidney excretion functions, leading to the reduction of kidneys' ability to completely excrete ammonia. This increases ammonia released into renal veins repeating the vicious cycle of high ammonia levels in the blood all over again.17 There is another argument that amino nitrogen required for synthesis of glutamate (essential for the conversion of ammonia to glutamine) is found in branched-chain amino acid (BCAA) from α-ketoglutarate in skeletal muscles.18 Therefore, continuous glutamate consumption allows catabolism of BCAA in the cytosol and attempts to compensate for it by influx from the extracellular fluid, which leads to further reduction in body BCAA. This explains BCAA deficiency in liver disease patients. Also, decreased levels of BCAA vis-à-vis increased levels of aromatic amino acids (phenylalanine, tyrosine, and tryptophan) in the central nervous system create an imbalance of dopamine, noradrenaline, and serotonin synthesis, which have been implicated in the formation of false neurotransmitters octopamine, phenylethanolamine, and tyramine.19 Acute liver failure, chronic liver failure, and cirrhosis are the main liver diseases that cause HE. Patients with high ammonia level show more rigorous symptoms, while relatively mild symptoms are seen with low serum ammonia levels.20 Hence, the correlation between severity of HE and ammonia level in plasma, cerebrospinal fluid, and the brain is also an indication that ammonia plays a vital role in etiology of HE.
Ammonia in the brain
Recent studies revealed that astrocyte cell membrane is highly permeable to ammonium ions; therefore, low concentrations of ammonia can permeate and cross the blood–brain barrier irrespective of systemic plasma ammonia level.21 However, excess ammonia in systemic blood increases the concentration entering the brain, which then triggers several automatic responses and mechanisms to reverse the influx. Glutamine synthetase in the brain is located mainly in astrocytes. There is neuron–glial metabolic interaction in the form of glutamate glutamine cycle, which keeps low concentrations of glutamate at all times and protects neurons from toxic effects of ammonia. The astrocytes, which protect neurons against excitotoxicity, convert the excess ammonia and glutamate into glutamine catalyzed by the enzyme glutamine synthetase. The glutamine formed is released back into extracellular space where it is used by neurons as glutamate precursor. Glutamate released by neurons can then be recaptured by astrocytes and converted into glutamine, thereby maintaining a proper balance of glutamine and glutamate levels.22 Unfortunately, alterations in glutamine synthetase expression affect the function of citric acid cycle in neurons and interfere with the energy levels of synaptic processes of the brain cells resulting in imbalance of nitrogen–energy homeostasis. Hyperammonemia induces astrocyte compensatory morphological changes, which may disrupt other activities such as neurotransmitter uptake, synthesis of glutamine synthetase, and blood–brain barrier transport. Hyperammonemia in the brain increases glutaminase activity in astroglial cells and thus leads to enhanced accumulation in astrocytes, resulting in astrocytosis, intracranial pressure, and cerebral edema making glutamate–glutamine cycle an essential step in brain edema. Researchers observed that inhibition of glutamine synthetase using methionine sulfoximine resulted in reduced ammonia-induced brain edema concluding that high glutamine level disrupts the normal cerebral osmotic regulatory functions.23 The glutamine produced in astrocytes in response to active glutamate receptors favors nitric oxide formation by activation of nitric oxide synthetase. Nitric oxide has been implicated in several central nervous system diseases. Another cause of hyperammonemia-induced brain edema is cytokine exudation, increase in lipopolysaccharides, and activation of nuclear factor kappa B as well as oxidative and nitrative stress. Experiment on rats revealed that there was a significant reduction in brain edema and astrocyte swelling when rats were treated with ammonia and nuclear factor kappa B inhibitor as compared with the effect when treated with only ammonia in case there was tremendous swelling.24 According to Kato et al., unlike the lumen of systemic blood vessels, endothelial cell linings of the brain microvessels are joined by tight junctions and have no fenestrations; therefore, exchange of substances takes place through specific carrier-mediated transporter.25 However, high concentration of ammonia in the brain increases endocytosis, vesicular tubule canalicular transport, and swelling of cerebral parenchyma cells with increase water content because of disturbance of cellular metabolism without any impairment of vascular permeability. This distorts the permeability of the blood–brain barrier with leakage of protein-rich edema fluid into the extracellular space, resulting in brain edema and disturbance of normal brain function.
Classification of hepatic encephalopathy
Hepatic encephalopathy is classified according to the underlying disease and the severity of manifestations. According to the 1998 World Congress in Vienna (Table 1), HE can be classified as type A, type B, and type C.26 Type A occurs because of acute liver failure; type B is caused by portal-systemic bypass without associated intrinsic liver disease; and type C occurs in patients with liver cirrhosis and portal hypertension or portal systemic shunts. Type C can be subclassified into episodic HE, persistent HE, and minimal HE. Episodic HE is seen in patients with delirium (an acute mental disturbance depicted by confusion, lack of attention, and incoherent speech without preexisting dementia). Persistent HE is typified by disturbance in alertness, inability to focus on task at work, social disintegration, and insomnia. Patients with minimal HE have no clinical symptoms, usually referred to as “subclinical encephalopathy.” Unlike the clinical symptomatic HE patients, minimal HE has no distinct character of brain dysfunction; therefore, its diagnosis requires sophisticated tools rather than the conventional method of elimination by exclusion.27 According to the West Haven Criteria, HE can be graded based on level of consciousness and semiquantitative mental status into grade 0, grade 1, grade 2, grade 3, and grade 4.28 Grade 0 is used when there is no abnormality detected using clinical signs and symptoms. Grade 1 is characterized by prodrome, lack of awareness, sleep disturbance, and shortened attention span. Grade 2 includes confusion, lethargy, muscle tone rigidity, and minimal disorientation of time or place. Grade 3 is a more advance stage with stupor, clonic spasm, but average response to voice and pain. Grade 4 is the final stage and the most severe; the hallmark is coma. There is total loss of consciousness and unresponsive to voice but may respond to pain (Table 2). The Glasgow Coma Scale is used to measure the level of consciousness of grade 4 patients according to the response to pain, eye opening, verbal behavior, and motor reflex. Depending on the degree of severity, HE can be classified as covert or overt.29 The covert type is also known as minimal hepatic encephalopathy (MHE). It is the earliest form and affects about 80% of patients with liver cirrhosis.
| HE type | Nomenclature | Subcategories |
|---|---|---|
| A | Encephalopathy associated with acute liver failure | — |
| B | Encephalopathy associated with portal systemic bypass without liver disease | — |
| C | Encephalopathy associated with cirrhosis and portal hypertension or portal systemic shunts | Episodic HE |
| Persistent HE | ||
| Minimal HE |
- HE, hepatic encephalopathy.
| Grade | Clinical features | EEG | Sonic criteria |
|---|---|---|---|
| 0 | Normal | Normal readings | Covert |
| MHE | Tremor, little changes in attention span and driving, and visual disturbance | Normal EEG results | Covert |
| 1 |
1. Prodrome, anxiety, personality, and behavioral changes 2. Change in sleep pattern, irritability, and depressed. There may be the presence of flapping tremor |
No significant readings. Normal or close to normal EEG result | Covert |
| 2 |
1. Confusion, lethargy, muscle rigidity, and disorientation of time and place 2. Drowsy but easily rousable and lethargic. Flapping tremor present |
Abnormal readings on the EEG | Overt |
| 3 |
1. Stupor and deep sleep but responds to pain and voice 2. Presence of flapping tremor, somnolence, confusion, and forgetfulness |
Abnormal readings on the EEG | Overt |
| 4 |
1. Coma, patient is unconscious 2. Does not respond to pain and voice |
Abnormal readings on the EEG | Overt |
- EEG, electroencephalogram; HE, hepatic encephalopathy; MHE, minimal hepatic encephalopathy.
Diagnosis of hepatic encephalopathy
- Exclude all other potential causes of encephalopathy,
- Identify and manage all precipitating factors, and
- Perform quick empirical treatment with 72-h monitoring.
If there is improvement in patient's condition, HE is confirmed; if not, alternate treatment plan or differential diagnosis should be considered.30 Diagnosis of HE typically depends on history, signs and symptoms, physical examination, and laboratory testing of blood ammonia. This method of diagnosis is considered the “gold standard”31; however, solely relying on this can be misleading as there are several cases of missed diagnosis (especially covert HE). Intracranial lesions and neurological diseases such as tumor, infections (encephalitis), stroke, and subdural hematoma can present similar symptoms as HE.32 Epilepsy, drug (narcotics/sedatives), or chronic alcohol intoxication, and neurological syndromes such as hepatocerebral degeneration, hepatic myelopathy, and neuronal dysfunction are all linked to liver insufficiency and must be ruled out.33 Recent development and standardization of more advanced and comprehensive tools for diagnosis has helped reduced cases of missed diagnosis. Some of these include psychometric hepatic encephalopathy score (PHES), neuropsychological tests, and inhibitory control tests.
Psychometric hepatic encephalopathy score test
Psychometric hepatic encephalopathy score measures the psychomotor speed, visual acuity, and attention span.34 PHES test consists of line tracing, serial dotting, digit symbol, digit span, canceling d-test, and number connection tests A and B.35 Psychometric driving simulator test helps in diagnosis of covert type of HE. Even though a good tool for diagnosis, comparing these seven test results of patients with MHE and healthy volunteers requires significant amount of time and for the fact that the test is ordered, administered, and interpreted by a psychologist make it not readily available.36 However, in the absence of PHES, the diagnosis of covert HE can be confirmed with two abnormal results from number connection test A, number connection test B, block design test, or digit symbol test.
Neuropsychological tests
Neuropsychological tests analyze the current mental status such as dementia, amnesia, anxiety, depression, and level of concentration.37 It also assesses the ability to do simple or complex calculations. Neuropsychological tests consist of critical flicker frequency, electroencephalogram (EEG), and evoked potentials.38
Critical flicker frequency measures the highest frequency at which a fused light (60 Hz or lower) that appears as a flicker can be distinguished by the brain and produces a feedback. The test is reliable, simple, sensitive, and easy to interpret. It can be administered without much expertise. Patients with worsening cognition typically have low results, and improvement after therapy produces higher results.39
Electroencephalogram measures the cortical cerebral activity of the brain.40 Patients with severe HE present abnormal patterns on EEG readings with slow rhythms (rhythm of alpha wake reduces and delta waves appear).41 The EEG is not a definite diagnostic tool but help recognize the decrease mean frequency of electrical activity of the brain and with other tests help elucidate diseases with similar clinical signs. Evoked potentials measure the time it takes for the brain to respond to a stimulus. It is characterized by the prolonged incubation of P300 in HE patients.42
Inhibitory control tests
Inhibitory control test records the attention span, response inhibition, and working memory.43 A great progress has been made to standardize the test for effective diagnosis of HE. Even though it requires higher-functioning patients, making it not a suitable and readily available, it can be administered by any trained healthcare professional rather than a psychologist.44
Radiological tests and neuroimaging
Radiological tests and neuroimagings are noninvasive tests aimed at ruling out lesions and to observe biochemical changes.45 There are no obvious observable pathological features of structural abnormalities at the gross anatomic level; however, current techniques, which include magnetic resonance imaging, positron emission tomography, and computer X-ray tomography, allow the evaluation of physiological and biochemical features of the brain.46 These tests must be performed in combination with other tests of sensitivity and specificity for reliable diagnosis.
Treatment of hepatic encephalopathy
In severe liver disease, many drugs can further impair cerebral function; therefore, careful selection of best treatment plan is a must. The exclusive use of drug in treatment of HE is the predominant option over the years. Most drugs target the reduction of ammonia and suppress the production of neurotoxin in the bowel.47 Few standard regimens exist for managing HE until now. Most countries rely on their own treatment guides and drugs approved by the Food and Drug Administration for the treatment of HE (Table 3). The most widely accepted drug as the first line of treatment is lactulose. Other drugs such as rifaximin and neomycin are also used. Dietary therapy, phenylbutyrate, BCAAs,48 probiotics, and other antibiotics are used on a case-by-case basis and not a routine treatment guide for every case. Targeting the cycle of glutamine conversion to ammonia and vice versa during treatment is also a laudable suggestion that needs to be further researched. Recent recommendation by the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases highly advises combination of lactulose with rifaximin as an add-on for prevention of recurrent episodes of HE after the second episode.49 Outcome of the use of nutritional interventions and other drugs undergoing clinical trials also showed positive results and highly recommended as adjunct options. Summarized in the succeeding texts is the mechanism of individual drugs and their possible advantages and disadvantages.
| Treatment plan and drug | Mode of action | Recommended time to use |
|---|---|---|
| Lactulose/lactitol | Reduces ammonia production by acidification of the colon, acts as laxative, and aids gut microbiome repair | First line of treatment in most countries |
| Rifaximin | Antibiotic with high efficacy and nonabsorbable and reduces ammonia production | Given to patients intolerant to lactulose or used in combination |
| Neomycin | Curbs the bacteria that produce ammonia in the gut. It is recommended for acute and overt patients but not for chronic use | As adjunct to lactulose and alternative for rifaximin |
| Metronidazole, flumazenil, vancomycin, and quinolones | These are poorly absorbed antibiotics used to decrease colon flora. Their side effects make them an undesirable option for long-term usage | Used in the absence of better option. Its neurotoxicity effect makes it not good for long term |
| l-Ornithine and l-aspartate | Synthetase for the OTC and CPS1 transamination in urea cycle as substrate for glutamate transamination | In addition to other drugs for more efficient result |
| Probiotics | Food supplements suitable for efficient bowel movement and balancing normal flora | Second and third line treatment option with other drugs |
| Laxatives | Produce bowel movement and relieve constipation | In combination to other drugs to speed up bowel movement |
| Branched-chain amino acid | Acts as substrate for protein and regulates nutrient deficiency | Used in combination with other drugs |
| Phenylbutyrate | Reduces ammonia in urea cycle disorders as an alternate pathway nitrogen excretion | Can be combined with other drugs especially in urea disorder |
| Diet | Adequate support of body mass, energy, and protein | Adjunct treatment option |
- Flumazenil is useful in improving mental status.
- CPS1, carbamoyl phosphate synthetase I; HE, hepatic encephalopathy; OTC, ornithine transcarbamylase.
Lactulose
The first line of treatment drugs is nonabsorbable disaccharides lactulose (or β-galactosidofructose) and lactitol (or β-galactosidosorbitol).50 They are the most widely used drugs generally characterized as osmotic laxatives. They are able to pass through the small intestine into the large intestine unchanged because the mucosa lining of the small intestine does not have the necessary enzymes to break these disaccharides into their small monosaccharide units. In the gut, they are broken down into lactic acid and acetic acid by bacteria in the colon. The acidification causes the environment to be less favorable to the ammonia-producing bacteria, which help convert ammonia to less absorbable ammonium and shift the normal flora in the colon from urease to nonurease-producing bacteria. It also has a laxative effect, increasing the rate of bowel movement, which removes intestinal nitrogen and produces excess ammonia in the body. Lactulose is given in doses of 25 mL to about 45 mL orally. The goal is to be able to have two to four bowel movements per day. The dose can be increased with severe form of HE. The long-term use of lactulose to minimize recurrences of HE has been shown to be effective in patients with cirrhosis and have had an episodes of HE. Lactulose also showed great efficacy as primary prophylaxis against the development of overt HE in patients with cirrhosis. There is no significant difference between lactulose and lactitol. Results of clinical trials using both drugs showed similar efficacy in both.51 However, there are concerns about the side effects such as nausea, diarrhea, flatulence, and bloating associated with the use of disaccharides. But because the dosages can be adjusted to minimize these side effects and also the advantages greatly outweigh the disadvantages, using them is still the main option available. Long-term use also helps develop tolerance to the side effects.52 Sole use of lactulose produced great result in lowering ammonia levels and improving cognitive function but could not prevent sepsis hence the recommendation to use it in combination with rifaximin (or other approved antibiotics).53
Rifaximin and other antibiotics
Another treatment option for HE is the use of antibiotics. Oral nonabsorbable antibiotics such as rifaximin or neomycin are either given to patients who are intolerant to lactulose or as a second line of treatment to complement existing therapy.54 These antibiotics curb the bacteria that produce ammonia in the colon and associated toxins by suppressing the intestinal flora. Rifaximin is a nonabsorbable derivative of rifamycin. Rifaximin covers a wide range of aerobic and anaerobic Gram-positive and Gram-negative bacteria. It is passed out in feces unchanged. Recent studies into the efficacy of neomycin showed some level of small absorption leading to ototoxic and nephritic side effects. This has caused doubt about the continuous use of neomycin as a suitable treatment of HE.55 The efficacy of rifaximin surpasses that of lactulose and neomycin, but it is not as cost-effective as lactulose in a long-term usage. The current recommendation on treatment suggested combination of rifaximin and lactulose.56 Other antibiotics such as metronidazole, oral vancomycin, paromomycin, flumazenil, and oral quinolones are also drugs of choice for the treatment of HE (though still undergoing clinical trials) and not approved or widely used because of their major side effects. Metronidazole and vancomycin need to be used under close supervision and regular monitoring as long-term use pose the risk of peripheral neuropathy.57
l-Ornithine and l-aspartate
l-Ornithine and l-aspartate (LOLA) are involved in the urea cycle as substrate for glutamate transamination. It is a synthetase for the transamination of ornithine transcarbamylase and carbamoyl phosphate. LOLA is a source of aspartate, which helps glutaminase to convert ammonia and accelerate its elimination from the body. LOLA used in the treatment of HE is a salt made of ornithine and aspartic acid. Patients treated with intravenous LOLA showed a great progress on their psychometric test, EEG readings, serum ammonia, and clinical signs.58 Although not widely used in many countries, the efficacy of LOLA coupled with low side effect makes it a viable option for patients with portosystemic shunt HE. Studies into the use of oral LOLA versus placebo showed significant improvement in the neurocognitive tests and ammonia level.59 The dosage of LOLA should be reduced in patients with concomitant renal disease. LOLA is only used in addition to other drugs and as such is good as adjuvant treatment but not a stand-alone option.
Probiotics
Probiotics are live microorganism food supplement, which, when given in adequate amounts, help balance the normal flora of the gut, producing a health benefit on the host.60 These are food supplements/ingredients that cannot be digested by the intestine and help stimulate bacteria in the colon, which are essential for nitrogen excretion. Random studies on patients produced an encouraging result including efficient bowel movement, lower serum ammonia levels, and improved psychometric test results.61 Probiotics, like the antibiotics, should be the second-line and third-line treatment options used in addition with nonabsorbable disaccharides.
Laxatives
Laxatives help increase bowel movement and relieve constipation.62 Most laxatives work by retaining water in the intestinal lumen, softening the stool, and lowering the pH. This action increases colonic peristalsis, and the low pH creates an environment uncomfortable for ammonia-producing bacteria. The efficacy of laxatives in treating HE is an ongoing debate; however, some reports suggested improvement in test results of patients who had recent episodes of HE. In a trial that compared the effects of laxatives versus placebo, once again, the result proved that laxatives reduce blood ammonia levels and reduced HE symptoms significantly compared with placebo. This notwithstanding, several more trials are needed to fully understand and accept their benefit into a routine treatment alternative plan.
Branched-chain amino acid
These are amino acids composing of valine, leucine, and isoleucine. Patients with liver failure have an imbalance between BCAA and aromatic amino acids. BCAA enhances ammonia detoxification to glutamine.63 Urea cycle defect reduces the conversion of ammonia to urea; however, oral BCAA improves metabolic health by promoting detoxification of ammonia in skeletal muscle.64 Recent trials in HE patients with or without symptoms of muscle wasting and who did not improve with traditional drugs but were given BCAA produced a relatively better result.65 However, adverse side effects such as cataplerosis, altered neurotransmission, and glutamine catabolism to ammonia may be associated with use of BCAAs in the treatment of cirrhosis.63 There is the need for new therapeutic strategies so as to enhance the effectiveness of BCAA and at the same time reduce the side effects.
Phenylbutyrate
Phenylbutyrate is used in the management of urea cycle disorders to reduce ammonia by providing an alternate pathway to urea for waste nitrogen excretion. Phenylbutyrate undergoes oxidation to form phenylacetate, which is conjugated with glutamine to form phenylacetylglutamine in the liver and the kidneys and excreted in urine.66 This reduces glutamine in the blood, temporarily suppressing ammonia production from glutamine, and promotes ammonia detoxification in the glutamine synthetase reaction. Recent random control trials using glycerol phenylbutyrate and sodium phenylbutyrate on patients who had two or more episodes of HE in the last 6 months indicated fewer recurrent episodes. Even though phenylbutyrate is well tolerated in adults with cirrhosis, it has been shown to decrease BCAA levels in blood plasma.67 This is probably due to enhanced glutamine depletion and removal from the body, which in turn activates BCAA catabolism. These results indicate that BCAA and glutamine supplementation are needed when phenylbutyrate is used therapeutically.68 Ornithine and phenyl acetate also help by converting ammonia to glutamine and subsequently forming phenylacetylglutamine, which is excreted in urine. Marked decrease in BCAA levels in blood plasma has been reported after phenylbutyrate treatment; therefore, its usage should be decided based on individual circumstances and completely avoided in urea cycle disorder patients.
Dietary therapy
Having a nutritionist help in selecting diet for patients with HE is a great and recommended approach. Restriction in amount of protein intake should only be advised based on expert review as this can lead to muscle wasting.69 Nutritional intervention is necessary for patients intolerant to the first line of treatment drugs or patients treated for couple of months with no improvement.70 Muscle tissue plays an important role in converting ammonia to glutamine, thereby reducing blood ammonia level; therefore, proper care should be taken to prevent malnutrition as this greatly affects the prognosis.71 Past suggestion of reducing protein intake in the hope of reducing ammonia level was later disproved as it leads to muscle and energy loss. Recent researches into the benefit of nutrition therapy indicated that daily recommended intake of energy should be between 35 to 40 kcal/kg and 1.2 to 1.5 g protein/kg. Regular caloric ingestion is essential to prevent unnecessary utilization of gluconeogenesis and keep constant visceral glucose output. This is especially important in patients with HE because conversion of amino acids for glucose production depletes tissue protein stores and produces ammonia. It is therefore important that patients avoid fasting for longer than 5 h during the daytime and so should be encouraged to take small, frequent meals or snacks in between meals evenly distributed throughout the day. Recent results from several reviews concluded that a late evening snack reverses the breakdown of aberrant substrate calories in patients with cirrhosis; therefore, HE patients should be encouraged to tailor their eating patterns along these recommended approaches.
Conclusions
Hepatic encephalopathy has a huge impact on the quality of life; therefore, immediate and total care is needed to prevent morbidity and mortality. MHE patients could go undiagnosed and are more prone to motor vehicle accidents; hence, the use of neuropsychometric test rather than solely relying on ammonia to diagnose is highly encouraged. Nonabsorbable disaccharides (lactulose) alone could not prevent sepsis; thus, the recommended therapy for long-term management is a combination of lactulose and rifaximin for prevention of recurrent episodes. Laboratory results of patients treated with the other treatment options discussed in this paper produced improved outcome and good prognosis; however, more clinical trials are necessary to evaluate their efficacy and draw conclusion on their routine usage.
Acknowledgments
This study was partly supported by the Scientific Projects of Zhejiang Province (2015C33148), Zhejiang Key Laboratory of Medicine and Pathophysiology (201703), and the KC Wong Magna Fund of Ningbo University.

