MLH1 Differential Allelic Expression in Mutation Carriers and Controls
Summary
Germline defects in the MLH1 gene are associated with Lynch syndrome. A substantial proportion of these mutations leads to premature termination codons and can induce nonsense mediated decay (NMD) of the corresponding transcript. Resulting allelic expression differences represent a fast and inexpensive method to identify patients carrying MLH1 mutations. In patients and controls, we show that allelic expression imbalance (AEI) can be readily detected in RNA extracted from whole blood from patients carrying mutations expected to elicit NMD using mass spectrometry. Mutations closer to the 5′ end of the gene tend to show smaller imbalances. AEI can also be detected in normal controls. Analysis of allelic expression in controls and individuals with mutations not expected to exhibit NMD revealed that MLH1 expression is influenced by sequence variation acting in cis. A maximum likelihood framework was used to identify two SNPs, rs1799977 (c.655G>A; p.I219V) and rs1800734 (c.-93 G>A) that are independently associated with expression. These influences are, however, small compared to the differences associated with pathological variants.
Introduction
Mutations in the mismatch repair (MMR) gene MLH1, are among the most frequent causes of Lynch syndrome (Hereditary Non-Polyposis Colon Cancer HNPCC, OMIM 120435)(Peltomaki & Vasen, 1997), an autosomal dominant syndrome that accounts for approximately 5% of all colorectal cancers (Bocker et al., 1999). Pathogenic variants in other components of the MMR system, MSH2, MSH6, and PMS2 account for most other cases. MLH1 and MSH2 are the most frequently implicated (Peltomaki & Vasen, 2004; Woods et al., 2007). Lifetime risk for mutation carriers of developing cancer was originally overestimated, raising the question of potential modifiers of penetrance and suggesting that a larger proportion of mutations in these MMR genes may be expected to occur in cases without a family history (Terdiman, 2005). This should lead to an increase in demand for mutation screening (Terdiman, 2005). The situation is complicated by the realization that a substantial proportion (30%) of mutations may not be detectable using routine sequencing (Casey et al., 2005), while epigenetic inactivation of one allele can cause cancer predisposition and may even be transmissible through the germline (Suter et al., 2004; Hitchins et al., 2007; Valle et al., 2007).
Approximately 50% of known mutations in MLH1 and MSH2 result in premature termination of the encoded proteins, and may trigger nonsense mediated decay (NMD), the preferential degradation of mRNA molecules containing a mutation that leads to premature termination (Weischenfeldt et al., 2005; Amrani et al., 2006; Behm-Ansmant & Izaurralde, 2006). Assessing clinical utility requires knowledge of the extent of physiological variation. Expression levels of MLH1 have been described to vary by a factor of four between quiescent and proliferating cells (Matheson & Hall, 2003; Iwanaga et al., 2004). This degree of variability could obscure pathological expression and motivated the examination of allelic expression differences as a diagnostic tool (Curia et al., 1999; Tournier et al., 2004). Relative transcript levels of alleles can be measured in heterozygotes for a transcribed SNP (allelic expression imbalance, AEI). Here we will use the “degree of imbalance” or “relative overexpression” to describe the ratio of the level of the highest expressed allele to that of the other allele. We use the term allelic expression ratio (AER) for the ratio of the allele carrying a certain residue at the transcribed SNP, divided by that of the other allele. AER is relatively robust since each allele acts as a parallel internal control for the other.
The efficiency of NMD is known to vary between tissues (Bateman et al., 2003), possibly between individuals (Resta et al., 2006) and between different mutations. In general, mutations leading to truncation less than 55 nucleotides upstream from the last exon/intron boundary or downstream from this junction or to premature termination within the first coding exon do not elicit NMD (Perrin-Vidoz et al., 2002; Tournier et al., 2004). Studies have examined only a small number of mutants (Curia et al., 1999; Tournier et al., 2004), and relied heavily on lymphoblastoid lines (Curia et al., 1999; Tournier et al., 2004). Data from RNA extracted from peripheral blood cells are relatively sparse
The degree of imbalance published in previous studies for MLH1 mutation carriers was less than threefold (Tournier et al., 2004). Recent work has shown that such a degree of imbalance can be observed in a variety of genes, often the result of sequence variation affecting the promoter (e.g., Yan et al., 2002; Heighway et al., 2003; Lo et al., 2003; Loeuillet et al., 2007; Miyamoto et al., 2007; Cheung et al., 2008). Indeed, allelic expression differences have been used to identify genes whose expression is likely to be affected by cis acting polymorphisms and to map the sites responsible (Teare et al., 2006). For MLH1, full transcriptional activity as determined by reporter assays has been achieved using a fragment containing the 300 bases upstream from the transcriptional start site (Quaresima et al., 2001) and several SNPs in this region have been described. However, elements affecting transcription in cis can be widely distributed throughout a locus and may be located in neighboring loci, an extreme example being the action of Xist (Brockdorff, 2002).
Allelic expression studies for MLH1 to date have used a limited number of samples; and no allelic expression differences in normal individuals have been reported. The aim of this study was to investigate the use of allelic expression as a diagnostic tool in MLH1 using RNA from peripheral blood lymphocytes (PBLs), to assess physiological variation of MLH1 allelic expression and to identify sites associated with any differences in expression levels.
Material and Methods
Material
Peripheral blood for DNA and RNA extraction was collected from 148 participants of the CAPP2 (Colorectal Adenoma/Carcinoma Prevention Program study Burn et al., 2008). This was a double-blind study aiming to assess the protective effects of resistant starch and enteric coated aspirin on a high-risk population. Patients included in CAPP2 had either mutations in an MMR gene or a family history that fulfilled the Amsterdam criteria (Wijnen et al., 1998). We also collected blood from 145 anonymous donors mainly from the North East of England participating in the “People of the British Isles” study. This study aimed to collect blood samples from rural populations throughout the British Isles, which will be used to look at the patterns of genetic variation around the United Kingdom. The cancer type as well as the MLH1 mutation status of these participants is unknown. Additionally, DNA was obtained for 37 MLH1 mutation carriers (representing 27 different mutations) and 37 individuals without history of colorectal cancer or known MLH1 mutations from the Scottish reference collection.
Ethical Approval
Samples were obtained under the ethical permissions granted for the CAPP2 clinical trial based on MMR mutation carriers, for the Scottish population based colon cancer collection and for the People of the British Isles, a large-scale study of normal variation.
RNA Extraction
RNA extraction was performed on 2.5 ml of blood, collected using the PAXgene system (Qiagen, Crawley, UK) and following the protocol recommended by the manufacturer without DNAse treatment and eluted into an 80-μl volume.
DNA Extraction
DNA was extracted from 700-μl blood using the MagAttract DNA blood midi M48 kit (Qiagen).
Reverse Transcription
Reverse transcription was carried out using the Superscript III first strand synthesis system (Invitrogen, Paisley, UK) and random primers.
Choice of Markers
Allelic expression was assessed using rs1799977 (I219V), in exon 8 of hMLH, (http://www.ncbi.nlm.nih.gov/projects/SNP) considered the most heterozygous coding SNP. We genotyped rs1800734 (often designated −93G/A) because of its potential to affect GT-IIB (GT-motif 2B) and NF-IL6 (interleukin-6-regulated nuclear factor) binding site in the MLH1 promoter (Ito et al., 1999). The remaining markers rs1540354 and rs4647222, were chosen using iHAP (Song et al., 2006) so that the proportion of haplotype diversity captured is above 95%.
Genotyping
A MassArray system from Sequenom (Sequenom, Hamburg, Germany) and homogenous Mass Extend chemistry were used for genotyping and determination of allelic expression by MALDI-ToF mass spectroscopy. The primers used are listed in Table S1. rs1799977 was amplified as a uniplex, and rs1800734, rs1540354, and rs4647222 as a multiplex. Mass spectra were analyzed using the MassARRAY Typer 3.01 software. Allelic expression ratios were estimated as the ratios of the area under the G and A peaks and were done in triplicate. Results from the amplification of genomic DNA were used as equimolar reference to normalize the cDNA values. Subsequent analyses used the logarithm of the ratio.
Analysis of Allelic Expression Ratios
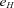 follows a lognormal distribution with
follows a lognormal distribution with 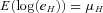 and
and 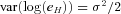 , where
, where  does not vary between different alleles. The log of the ratio between the expression levels of both alleles, I, can therefore be assumed to be normally distributed,
does not vary between different alleles. The log of the ratio between the expression levels of both alleles, I, can therefore be assumed to be normally distributed, 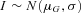 where the mean
where the mean 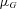 depends on the genotype
depends on the genotype 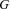 . We will model the expected value as a linear combination of the influences of the typed polymorphisms, that is, assume that the polymorphisms affect expression in a multiplicative manner:
. We will model the expected value as a linear combination of the influences of the typed polymorphisms, that is, assume that the polymorphisms affect expression in a multiplicative manner:

 is a parameter quantifying effect of the ith cis acting polymorphism on expression, and
is a parameter quantifying effect of the ith cis acting polymorphism on expression, and 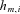 characterizes the phase between the transcribed marker m and the cis acting polymorphism
characterizes the phase between the transcribed marker m and the cis acting polymorphism  . If we arbitrarily designate for each marker one of the alleles 0 and the other with 1, then
. If we arbitrarily designate for each marker one of the alleles 0 and the other with 1, then 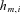 is defined as:
is defined as:


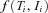 designates the density for an individual with the (phase-unknown) genotype
designates the density for an individual with the (phase-unknown) genotype 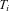 and an expression ratio
and an expression ratio 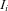 and the index
and the index  runs through all individuals included in the study. For an individual
runs through all individuals included in the study. For an individual 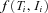 can be written as
can be written as

 is the probability of observing the phase-unknown genotype Ti, conditional on phase-known genotype G, and
is the probability of observing the phase-unknown genotype Ti, conditional on phase-known genotype G, and 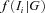 the density of observing and given the genotype G.
the density of observing and given the genotype G. 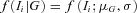 and
and 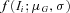 denote the density of a normal distribution with the individual expression ratio
denote the density of a normal distribution with the individual expression ratio 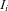 as variate, the genotype-dependent mean
as variate, the genotype-dependent mean 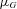 and the variance
and the variance 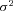 . Therefore
. Therefore  depends on
depends on 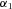 ,
,  , and on the genotype frequencies
, and on the genotype frequencies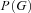 . However, in general, there will be some individuals, such as those that are homozygous for the transcribed marker, for whom no information on allelic expression is available. Therefore our sample can be subdivided into two disjoint sets, one consisting of individuals for whom AER has been measured and a second for whom it has not. The likelihood for the whole sample can be represented as the product of two components.
. However, in general, there will be some individuals, such as those that are homozygous for the transcribed marker, for whom no information on allelic expression is available. Therefore our sample can be subdivided into two disjoint sets, one consisting of individuals for whom AER has been measured and a second for whom it has not. The likelihood for the whole sample can be represented as the product of two components.

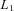 represents the individuals for whom AER has been measured and has been described above, and
represents the individuals for whom AER has been measured and has been described above, and 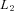 represents the individuals for whom only the genotype is available.
represents the individuals for whom only the genotype is available.
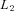 can be represented as
can be represented as


 as a problem with incomplete data that is solved using an Expectation Maximization (EM) procedure. In general, the likelihood will depend on
as a problem with incomplete data that is solved using an Expectation Maximization (EM) procedure. In general, the likelihood will depend on 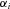 ,
,  , and on the genotype frequencies
, and on the genotype frequencies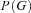 , allowing the testing of specific hypotheses as likelihood ratio tests.
, allowing the testing of specific hypotheses as likelihood ratio tests.Other Statistical Analyses
Twenty-two mutations were present in more than one individual and in 16 cases allelic expression allowed us to determine which of the rs1799977 alleles was in phase with the mutation. This information was used to estimate the frequencies of the “mutation-carrying” and “wild-type” haplotypes. Familial relationships were not available for most cases. We assumed that individuals with the same mutation shared one haplotype by descent. Thus a group of n individuals with the same mutation can be represented by n+ 1 haplotypes, that is, the “mutation” haplotype and n wild-type haplotypes. The likelihood of the haplotype distribution among mutation carriers can be represented as a function of the frequency of mutation carrying haplotypes Hm and of the wild-type haplotypes Hw: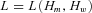 . We estimated the haplotype frequencies using an EM procedure and compared the hypothesis
. We estimated the haplotype frequencies using an EM procedure and compared the hypothesis 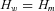 versus
versus 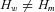 with a likelihood ratio test. The EM procedure was implemented in R (http://cran.r-project.org/). It also estimates the probability that each of an individual's possible haplotypes is wild-type or carries the mutation. All four SNPs were used for haplotype inference.
with a likelihood ratio test. The EM procedure was implemented in R (http://cran.r-project.org/). It also estimates the probability that each of an individual's possible haplotypes is wild-type or carries the mutation. All four SNPs were used for haplotype inference.
Criteria for Predicting Mutations Causing NMD
Following the conclusion from previous studies on AEI in individuals with truncating mutations (Perrin-Vidoz et al., 2002; Tournier et al., 2004) we designated mutations that lead to premature termination of the coded peptide downstream from the first coding exon and at least 55 bp upstream of the last intron/exon boundary, as predicted to cause NMD.
Results
From 66 individuals carrying 39 different MLH1 mutations, 36 had changes predicted to cause NMD, which represented 12 different mutations. From the 66 individuals, 29 were heterozygous for rs1799977 (c.655G>A SNP). AEI was measured in all 16 individuals heterozygous for this SNP who carried mutations predicted to cause NMD, representing nine different mutations (Table 1).
| Total | rs1799977 heterozygous | MLH1 NMD expected | AER measured | |
|---|---|---|---|---|
| Mutation carriers | ||||
| MLH1 | 66 | 29 | 36 | 27 |
| MSH2 | 52 | 13 | 0 | 9 |
| MSH6 | 2 | 1 | 0 | 1 |
| Unknown mutation | 28 | 11 | 0 | 5 |
| Controls | 145 | 70 | ? | 59 |
Figure 1 presents the allelic expression ratios; all groups showed a large variation in the extent of allelic imbalance. Among individuals predicted to have NMD, there were four overexpressing the G allele and 12 the A allele. If the samples represent 11 different mutations (see below), and assuming each mutation occurred only once, the distribution of two mutations having occurred in a chromosome carrying a G and nine in a chromosome carrying an A allele at rs1799977 is consistent with the frequency of the rs1799977 A allele observed among unaffected controls (0.69, P= 0.52, consistent with no preference for mutations to occur in a particular allele, see also Table S2). The degree of expression imbalance (Fig. 2), ranged from 1.42:1 to 2.72:1, for individuals carrying MLH1 mutations that should not cause NMD from 1.01:1 to 1.15:1 and for individuals with mutations in other MMR genes from 1.01:1 to 1.26:1. In the control group, the sample marked by an asterisk in Figure 1 has a value of 2.17:1. The range for the remaining samples from this control group is 1:1–1.37:1. Thus, with the exception of one value, there is no overlap between the AER from patients expected to show NMD and those from the other groups. However, even excluding the outlier in the control group, the difference can be small (1.42:1 compared to 1.37:1). There are no significant differences in the degree of imbalance between the control group and samples with MLH1 mutations not expected to cause NMD (Mann–Whitney test, P= 0.87), where mutations in other MMR genes were found (P= 0.36), or from patients where so far no mutation has been detected (P= 0.25).

Allelic expression ratios in samples from patients and controls. Represented is the ratio of the signal originating from the A allele at rs1799977 divided by that originating from the G allele. The error bars represent the standard error as estimated from three replicates.

Relative overexpression in samples from patients and controls. Represented is, for each individual, the ratio of the signals from the allele with the highest expression signal divided by that from the other allele.
Three mutations causing NMD were present in more than one individual (c.105_106insA in two individuals, c.901C>T in three patients, and c.1553_1554insA in five individuals). Figure 3 presents the AERs for these individuals. There is a significant association between AER and the mutation (multiple R2= 0.97, P= 3 × 10−6). However, this result reflects two factors: the degree of AEI and whether the mutation occurs on a chromosome carrying a G or an A allele at rs1799977. In the absence of other factors influencing allelic mRNA levels, the degree of imbalance will reflect the extent to which the mutant mRNA is degraded compared to that of wild-type mRNA. Analysis of variance reveals that for these three mutations, there was a significant association between the mutation and the degree of imbalance (multiple R2= 0.69, P= 0.017). We then investigated whether the extent of NMD varies according to the position of the mutation within the gene. We analyzed the correlation between the degree of imbalance and the distance between the mutation and transcription initiation site and found a significant correlation (R2= 0.64, P= 0.006). Next, we extended this analysis to all patients with MLH1 mutations expected to show NMD (Fig. 4). For this group, there was a significant correlation between the distance from the transcription start site and the degree of imbalance (R2= 0.45, P= 0.004; rank correlation test P-value = 0.006). Inspection of Figure 4 suggests that this trend may describe the effects of mutations toward the 5′ end of the gene, but we lack sufficient data to assess changes in the middle or in the 3′ region. There was no significant correlation between position and imbalance for mutations not expected to elicit NMD (P= 0.46).

Allelic expression ratios in individuals that share an MLH1 mutation. Represented are the carriers of the three mutations that were present in more than one individual in our study.

Relative overexpression among MLH1 mutation carriers. The values are plotted according to the genomic location of the MLH1 mutation in the corresponding individual. The triangle indicates the location of the transcribed SNP (rs1799977).
To assess whether “non-NMD” variation reflects the action of polymorphisms influencing expression, we investigated the association between haplotypes and allelic expression. We used an approach similar to forward stepwise regression: We first assessed the association between allelic expression ratios and haplotypes defined by one SNP. Once we identified the SNP providing the best fit, we analyzed the haplotypes defined by two SNPs; the one identified in the previous step and one of the remaining ones. This allowed identification of the two SNP combination that best fitted our observation. The process of adding one SNP at a time continued, until further addition failed to show significant improvement (Table 2). Column 3 of Table 2 indicates that, at the 1% level, there is a significant association between three of the SNPs analyzed and allelic expression. We took rs1799977, the SNP with the strongest indication of an association and asked whether including a second SNP in the haplotype led to a significantly better fit. The fourth column indicates that a model including rs1800734 in the haplotype leads to a significantly better fit. As shown in the fifth column the inclusion of additional markers to the rs1799977 and rs1800734 combination does not lead to a significant improvement. Since rs1800734 and the transcribed polymorphism are in strong disequilibrium (|D′| = 0.94), its effect can be visualized by plotting the allelic expression ratios for different rs1800734 genotypes. This is shown in Figure S1. There is a significant difference between the ratios for homozygous and heterozygous (P= 7 × 10−4 Mann–Whitney Test), suggesting an association between rs1800734 and allelic expression.
| Marker | Location1 | Effect included in the model | |||||
|---|---|---|---|---|---|---|---|
| 2 | rs1799977 | rs1799977rs1800734 | |||||
| L3 | P (1df) | L | P (1df) | L | P (1df) | ||
| rs1800734 | Promoter (−93) | −539.9 | 0.23 | −515.3 | 1.3 × 10−3 | - | - |
| rs1540354 | Intron 3 (7981) | −536.0 | 2.5 × 10−3 | −519.9 | 0.35 | −515.3 | 0.98 |
| rs4647222 | Intron 3 (9510) | −525.3 | 3.2 × 10−8 | −520.3 | 0.76 | −513.8 | 0.08 |
| rs1799977 | Exon 8 (18589) | −520.4 | 2.1 × 10−10 | - | - | - | - |
- 1The position with respect to the major transcriptional start site (Ito et al., 1999), is given in brackets.
- 2The log likelihood for the model that assumes no association between any SNP and AER is −540.6,
- 3Log likelihood.
We next examined whether the distribution of the haplotypes defined by rs1799977 and rs1800734 differed between mutation carrying and not carrying chromosomes. Analysis of our original set of samples (64 mutation carriers representing 39 different mutations) showed a trend with a P value of 0.09. We decided to extend our sample size and included samples obtained from Edinburgh (37 mutation carriers representing 27 different mutations). On its own, this group also showed borderline significance with a P value of 0.10. Analysis of the combined set resulted in a P value of 0.19. The estimated frequencies for the combined data set are presented in Table S2.
Discussion
Figures 1 and 2 show that, although mutations expected to cause NMD show a large degree of imbalance, there is also a substantial background variation between individuals. In this study, we relied on mass spectroscopy to analyze allelic expression. This method has been used to quantitate relative allele frequencies in pooled DNA and over the past 5 years it has extensively been used to assess AEI (e.g., Ding & Cantor, 2003; Knight et al., 2003; Knight et al., 2004; Gimelbrant et al., 2007; Lo et al., 2007). Compared to previous studies, that used real-time PCR (Chen et al., 2008), or gel electrophoresis (Curia et al., 1999; Montagna et al., 2002; Perrin-Vidoz et al., 2002; Tournier et al., 2004), mass spectroscopy can potentially analyze several genes simultaneously in a large number of samples at a low cost. Such an assay could complement sequencing strategies since it allows the assessment of changes outside commonly sequenced regions such as promoter or enhancer elements and could perhaps help to identify changes affecting expression where no DNA sequence alterations have been found (“epimutations”; Hitchins & Ward, 2009). The degree of imbalance we observed in carriers of MLH1 mutations predicted to cause NMD is consistent with that described in the study by Tournier et al. (2004). They observed ratios between 1.5:1 and 2.2:1 in peripheral blood lymphocytes, and one value from 4.2:1 in a lymphoblastoid cell line. Our results show that the level of imbalance is substantially larger in samples expected to show NMD than in samples with no known MLH1 mutation and anonymous controls. However, the magnitude of the imbalance in the presence of NMD is not particularly high compared to the physiological variation observed in other genes. The largest imbalance we observe in a mutation carrier is 2.7:1 (see Fig. 1) while a survey of normal tissues using microarrays found that 15% of all transcripts showed an imbalance of at least 4:1(Lo et al., 2003).
Among samples showing NMD, there is a tendency for the rs1799977 A allele to be under-expressed (see Fig. 1). Such an apparent preferential under-expression of one of the transcribed alleles in heterozygous individuals has recently been reported in BRCA1 and BRCA2 mutation carriers and can be interpreted as an increased risk of a mutation in individuals under-expressing one allele (Chen et al., 2008). Our observations suggest that, for MLH1, this overrepresentation simply reflects the lack of a strong bias for a mutation to occur in a particular background haplotype (see Results section). In this case, the probability of a certain allele carrying a mutation, or being subjected to NMD, will be proportional to its population frequency. Among the controls there was one sample that showed imbalance to an extent similar to that observed for mutations causing NMD. For ethical considerations, we were unable to sequence MLH1 in this sample. The probability of observing at least one mutant allele among 59 samples is 2.9% assuming that half the MLH1 mutations lead to NMD and that the frequency of mutant alleles in the populations is 1:2000. However it is possible that previous contact with a genetic diagnostic service could increase the willingness of an individual to participate in a scientific study and therefore this individual may be a MLH1 mutation carrier. An alternative explanation is that there are low-frequency alleles in the population that have a large influence on expression. The existence of such alleles would be interesting because their low frequency would suggest that they are under negative selective pressure.
Our data indicate that there are substantial differences in the ability of different mutations to cause NMD. In our samples, the extent of imbalance correlates with the position of the mutation with respect to the transcriptional initiation site. This suggests that MLH1 mutations close to the transcriptional initiation site have a smaller effect on mRNA levels than those more toward the middle of the gene. We did not observe AEI for mutations causing premature termination in the first or last exon. This is consistent with previous reports (Perrin-Vidoz et al., 2002; Tournier et al., 2004). Our results also show that allelic expression is not altered in individuals carrying mutations not expected to cause NMD. This contrasts with the findings from Curia et al. (1999) who reported up to 90-fold imbalances in samples with missense changes in MLH1 (Curia et al., 1999). In the absence of direct experimental manipulation, interpretation of AEI as the effect of a mutation in a specific gene requires understanding of the degree of variability in the normal population. For example, analysis of BRCA2 allelic expression in lymphoblastoid cell lines indicates that the degree of allelic imbalance in cell lines without a BRCA2 mutation can be similar to that observed for certain mutations expected to cause NMD (Montagna et al., 2002; Chen et al., 2008). For MLH1, we also find differences in allelic expression among individuals where no NMD is expected, however these differences are small. Such differences can be due to SNPs acting in cis on expression. Although the imbalances are small (see Fig. 1) the large number of samples allowed us to investigate whether there was an association between AEI and polymorphic sites in MLH1.
We present a new method to assess sites associated with allelic expression. This method represents a generalization of our previously published “two-marker” approach. It allows us to analyze a number of markers simultaneously. Samples homozygous for rs1799977 are included in the analysis that improves the ability to estimate the haplotype structure. This may be of interest in cases where there is a large degree of disequilibrium between markers and several markers may appear to be associated with allelic expression, or when the effect of one marker tends to mask the effect of another.
We used a stepwise procedure to identify markers associated with allelic expression. This led to the identification of two SNPs; the first, rs1799977, is the transcribed polymorphism itself. This is quite a common situation and several studies (e.g., Yan et al., 2002; Lo et al., 2003) have found a systematic overexpression of one of the transcribed alleles. This suggests that these SNPs can be used as markers for expression levels although they themselves may not affect transcription or mRNA stability directly, and their association probably reflects the extent of linkage disequilibrium across the genome. The second SNP, rs1800734, is located in the promoter region. This is also a common finding and reflects perhaps the high density of binding sites for transcription factors in promoters. This polymorphism has been the focus of considerable interest (e.g., Chen et al., 2007; Raptis et al., 2007; Allan et al., 2008), and has recently been shown to influence transcription using luciferase reporter assays (Mei et al., 2010).
We show a relationship between rs1800734 and expression in vivo. Compared to other studies, the effects of variation upon expression is modest, but since AEI can vary between tissues (Lo et al., 2003; Wilkins et al., 2007), this variation may have phenotypic consequences. A proportion of colorectal tumors, in particular those showing high microsatellite instability (MSI) lack MLH1 expression (Thibodeau et al., 1998; Jensen et al., 2008). This is believed to be predominantly a consequence of methylation of the promoter region (Kane et al., 1997; Furukawa et al., 2002). However transcriptional silencing of a DNA region can precede methylation (Hertz et al., 1999; Brockdorff, 2002), raising the general question whether differences in transcription can influence the susceptibility to silencing or more specifically whether variation influencing expression can also modify the disease phenotype.
For rs1799977, Kim et al. (2004) described a correlation between the constitutional rs1799977 genotype and MLH1 protein expression in colorectal tumors, while more recently Chen et al. (2007) found a direct association between promoter methylation and rs1800734 (c.-93A>G). In that study rs1799977 had the second lowest P-value, but with P= 0.07, the association to promoter methylation failed to reach statistical significance. We did not find an association between these two polymorphisms and disease onset in mutation carriers. This is perhaps not surprising given the small size of our sample at 101 individuals. The small sample size also precluded a more sophisticated analysis. For both polymorphisms, there have been partially conflicting reports regarding the association to the risk of different types of cancer (Krajinovic et al., 2002; Park et al., 2004; Lee et al., 2005; Beiner et al., 2006; Landi et al., 2006; Mei et al., 2006; Yu et al., 2006; An et al., 2008; Harley et al., 2008). However recent work suggests an association between rs1800734 and colorectal cancer risk (Chen et al., 2007), in particular for tumors showing MSI (Raptis et al., 2007) or lacking MLH1 expression (Allan et al., 2008).
Acknowledgements
We would like to thank the CAPP2 participants who contributed to this study and the participants of the “People of the British Isles” study from the North East. This work was supported by the UK Medical Research Council and by the Special Trustees Fund of the Newcastle University Hospitals. MGD and NC are supported by grants from Cancer Research UK (C348/A8896) and CORE (http://www.corecharity.org.uk).