

Plasma clearance
Abstract
Plasma (total, systemic…) clearance is determined by all the individual metabolizing/eliminating organ clearances and involves mainly liver and kidney clearances. Plasma clearance (a volume per time, i.e. a flow) expresses the overall ability of the body to eliminate a drug by scaling the drug elimination rate (amount per time) by the corresponding plasma concentration level. The interpretation of plasma clearance and inter-species comparisons are made easier by computing the overall body extraction ratio (from 0 to 1), which is the ratio of the body clearance divided by cardiac output. Plasma clearance is the most important pharmacokinetic parameter because it is the only one which controls the overall drug exposure (for a given bioavailability) and it is the parameter which allows computation of the dosage required to maintain an average steady-state plasma concentration.
Introduction
Plasma clearance is the most important of all pharmacokinetic (PK) parameters. It is almost systematically reported in PK papers but is only rarely actually used and almost never interpreted.
This review aims to explain what clearance is, why plasma clearance is the most important PK parameter, how to compute it, how to provide a physiological interpretation to its numerical value in domestic species, and what its main applications are in veterinary pharmacology. An authoritative review on this topic has been written by Wilkinson (1987). The descriptions in veterinary text books by Baggot (1977) and Riviere (1999) also provide good accounts.
The terminology ‘plasma clearance’: a preliminary comment
Plasma clearance is the term generally used, because plasma is the matrix most frequently used to measure circulating drug concentrations. When blood concentration is quantified, a blood clearance is computed. Blood and plasma clearances have the same numerical value as long as whole blood and plasma concentrations are equal. Whenever blood and plasma concentrations are different, attention should be paid to selection of the most appropriate matrix to evaluate the body capacity to eliminate a drug, and on the physiological interpretation of the calculated clearance (vide infra).
The plasma clearance measuring the overall ability of the living organism to eliminate a drug, is also termed total, systemic or body clearance. Unless stated otherwise, we will use all these terms as synonymous.
Why plasma clearance is the most important PK parameter
Plasma clearance is the most important PK parameter because it is one of the three determinants of a dosage rate. Figure 1 gives a pictorial view of the position of plasma clearance in relation to the dosing rate.

A fundamental relationship in steady-state conditions. Under steady-state conditions (for a multiple dosage regimen), the drug rate in should be equal to the drug rate out. This guarantees maintenance of a constant plasma (blood) drug concentration which in turn guarantees maintenance of the expected (steady-state) effect. Drug is considered by the animal as a foreign compound and is removed by the animal's eliminatory processes with variable efficiency. Clearance is the parameter which expresses the ability of the animal to eliminate a drug. Thus, the dosage regimen (which is under the clinician's control) should be proportional to clearance.
 ()
()Inspection of Eqn 1 shows that a dose is not a genuine drug parameter but rather a PK/PD variable, because it is influenced by a PK parameter (plasma clearance), a PK variable (bioavailability for a particular route of administration and a given formulation), and a pharmacodynamic (PD) parameter (target therapeutic concentration) which reflects the in vivo drug potency (for more explanation see Toutain, 2002).
What is plasma clearance?
 ()
() ()
() ()
()
Clearance: introduction of the concept using a single in vitro system. Assume an in vitro system in which there is an eliminatory mechanism [here cells suspended in a bathing drug (dot) solution]. Drug concentration in the solution is the driving concentration which supplies an unlimited amount of drug to cells. Here the drug concentration is considered as a constant because the amount of drug eliminated per unit of time (dx/dt), i.e. uptake by the cells is negligible in relation to the total drug amount in the beaker. The definition of clearance for this in vitro system is: 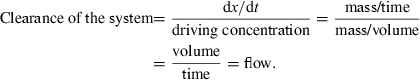 As there is a nonlimitation of the drug supply to the clearing organs, this clearance is termed ‘intrinsic clearance’ of the system (Clint). Clint is a measure of cellular drug removal, and is related to the Michaelis–Menten kinetic parameter according to the equation: Clint=Vmax/(KM+C), where Vmax is the maximal capacity of the system to eliminate the drug and KM, the concentration corresponding to Vmax/2. For the majority of drugs, KM (a concentration which is a measure of drug affinity for the clearing system (e.g. enzymes)), is much higher than C, and Clint (Vmax/KM) is a constant (parameter). The dimensional analysis of the expression of clearance indicates that Clint is numerically equal to the (virtual) volume of the system that is totally cleared of drug by unit of time i.e. a flow.
As there is a nonlimitation of the drug supply to the clearing organs, this clearance is termed ‘intrinsic clearance’ of the system (Clint). Clint is a measure of cellular drug removal, and is related to the Michaelis–Menten kinetic parameter according to the equation: Clint=Vmax/(KM+C), where Vmax is the maximal capacity of the system to eliminate the drug and KM, the concentration corresponding to Vmax/2. For the majority of drugs, KM (a concentration which is a measure of drug affinity for the clearing system (e.g. enzymes)), is much higher than C, and Clint (Vmax/KM) is a constant (parameter). The dimensional analysis of the expression of clearance indicates that Clint is numerically equal to the (virtual) volume of the system that is totally cleared of drug by unit of time i.e. a flow.
The rationale for scaling (or normalizing) the total rate of drug body elimination by the corresponding plasma concentration is to express the ability of the body to eliminate a drug by a genuine parameter (a constant), rather than by an excretion rate which is a concentration-dependent variable for a drug following a first-order elimination process. If drug clearance does not obey a first-order elimination process, clearance is no longer a parameter but instead becomes a concentration-dependent variable.
 ()
() ()
()How to evaluate plasma clearance
 ()
()
Estimation of plasma clearance: bolus vs. infusion study. Plasma clearance can be evaluated using different modalities of the i.v. administration namely, a bolus, short infusion, or infusion to reach steady-state conditions. This figure shows these approaches for a total administered dose of 100 mg/kg. (a) Parameters of a bi-compartmental model used to simulate data. (b) The i.v. bolus is the most convenient modality of administration. Clearance can easily be obtained either by the noncompartmental approach (Dose/AUC0--∞) or after modelling (Cl = K10 × Vc). (c) If for safety reasons an instantaneous bolus administration is dangerous, an alternative is a relatively short infusion (e.g.: 3 h allowing a reduction in the maximum plasma concentration (57.4 vs. 230 mg/L for the short infusion and the bolus administration respectively). Here also, the clearance can be obtained either by modelling or by a noncompartmental approach. (d) The last strategy consists of achieving a steady-state condition with a 48-h infusion (here infusion of 10 times the terminal half-life, which is of 4.62 h). Using a snapshot plasma concentration during the steady-state, clearance (0.333 L/kg/h) can be obtained with dose/infusion time = 2.08 mg/h/kg divided by Css (6.25 mg/L).
 ()
() ()
() ()
() ()
()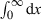 is equal to the total amount of drug ultimately eliminated, i.e. to the administered dose after i.v. and
is equal to the total amount of drug ultimately eliminated, i.e. to the administered dose after i.v. and 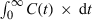 is the total AUC, therefore, Eqn 11 is equal to Eqn 7.
is the total AUC, therefore, Eqn 11 is equal to Eqn 7. ()
()Regardless of the approach selected, the total dose considered in Eqn 7, or the infusion rate in Eqn 12 should give the amount of drug which actually gains access to the systemic (arterial) circulation. If a fraction of the i.v. dose is destroyed by a first-pass pulmonary effect before reaching the arterial circulation (vide infra), or if the administered agent is a pro-drug (e.g. an ester for corticosteroids) that requires metabolic transformation (e.g. hydrolysis), estimation of total clearance by Eqn 7 or 12 can be very misleading. Figure 4 illustrates the example of methylprednisolone, for which clearance has been calculated by injection of either methylprednisolone itself or methylprednisolone sodium succinate.

Plasma clearance of methylprednisolone in the dog. Methylprednisolone (MP) is a nonhydrosoluble steroid which was administered by the i.v. route using a hydrosoluble salt of a succinate ester (methylprednisolone sodium succinate, MPS). After an i.v. administration of MPS, its concentration decreases rapidly giving the active moiety, i.e. MP. Using the MP plasma concentration profile to estimate the MP clearance would lead to gross overestimation of the true MP clearance, because only 44% of the MPS is transformed into MP. The true clearance of MP was obtained by administered MP itself by i.v. route (Toutain et al., 1986).
How to evaluate plasma clearance when it is not possible to administer the drug by the i.v. route
 ()
() ()
() ()
()In the same circumstance (i.e. when an excretion rate is directly and sequentially measurable), clearance can be evaluated by dividing the average excretion rate (Δx/Δt) by the plasma drug concentration at the time corresponding to the midpoint of the excreta collection period. Indeed, Eqn 3 indicates that clearance is the slope of the regression line linking the drug excretion rate to the plasma concentration. The advantage of this method is that it takes account of possible nonlinearity (Fig. 5).

Estimation of a clearance from experimental measurement of the excretion rate. Clearance can be computed from the ratio of the excretion rate (urinary, faecal, milk…) to the drug concentration in plasma (see Eqn 3). In practice, clearance is estimated by plotting excretion rate vs. drug concentration in plasma at the times corresponding to the mid-points of the matrix (urine, milk, faeces…) collection periods. The slope of the excretion rate vs. plasma concentration plot is equal to the clearance. If the drug is totally eliminated by the investigated matrix, the computed clearance is numerically equal to the plasma clearance. An advantage of this method is that it recognizes possible nonlinearity (e.g. for high plasma concentrations for a saturable elimination process).
The physiological interpretation of plasma (blood) clearance
Is a given computed plasma clearance a high, medium or low body clearance? This very relevant question is seldom addressed in veterinary papers because the answer is not straightforward. For instance, a plasma clearance of 20 mL/kg/min should be considered as high in cattle and horses, but rather low in rodents.
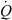 ) and an extraction ratio (E):
) and an extraction ratio (E):
 ()
()
Minimal model for an in vivo clearance. The grey box represents an organ or a system (several organs) capable of eliminating a drug. The organ is perfused with a blood flow (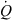 ). The input drug concentration is Cin. This is the driving blood concentration. The output concentration is Cout. If some drug has been eliminated during passage through the system, then Cout < Cin. The total amount of drug which is eliminated per unit of time can be obtained by mass balance consideration. It is given by the difference between the rate of drug presentation by arterial blood (
). The input drug concentration is Cin. This is the driving blood concentration. The output concentration is Cout. If some drug has been eliminated during passage through the system, then Cout < Cin. The total amount of drug which is eliminated per unit of time can be obtained by mass balance consideration. It is given by the difference between the rate of drug presentation by arterial blood (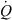 × Cin) and the rate of the drug leaving the organ in the (venous) blood, (
× Cin) and the rate of the drug leaving the organ in the (venous) blood, (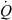 × Cout) i.e.
× Cout) i.e. 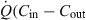 ), which is the rate of drug extraction. Applying to this simple system the general definition of clearance (see Eqn 2 in the text), allows expression of the clearance of the system as:
), which is the rate of drug extraction. Applying to this simple system the general definition of clearance (see Eqn 2 in the text), allows expression of the clearance of the system as: 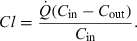 The ratio (Cin−Cout)/Cin is termed the extraction ratio (E), therefore, any in vivo clearance can be expressed as the product of a blood flow (
The ratio (Cin−Cout)/Cin is termed the extraction ratio (E), therefore, any in vivo clearance can be expressed as the product of a blood flow (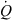 ) and an extraction ratio (E), a figure without dimension and with a numerical value between 0 and 1; hence a clearance is numerically equal to the volume of the entering blood from which drug appears to be totally extracted.
) and an extraction ratio (E), a figure without dimension and with a numerical value between 0 and 1; hence a clearance is numerically equal to the volume of the entering blood from which drug appears to be totally extracted.
 ()
() ()
()
A minimal physiological model for total body clearance. The entire body is considered as the drug reservoir. In mammals, kidney and liver are generally the two main organs for drug metabolism and excretion. Kidney and liver are the two parallel components of the clearing system. As a whole, the clearing organs are perfused by a blood flow equal to cardiac output (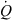 ). The model assumes that no drug is eliminated by lung (not represented) and that blood concentration is equal to plasma concentration. Under these conditions Eqn 13 applies (Cltot = Clrenal + Clhepatic + Clothers). By applying Eqn 4 to the system, with Cin being the arterial blood concentration (driving concentration) and Cout (the venous blood concentration), it can easily be demonstrated that
). The model assumes that no drug is eliminated by lung (not represented) and that blood concentration is equal to plasma concentration. Under these conditions Eqn 13 applies (Cltot = Clrenal + Clhepatic + Clothers). By applying Eqn 4 to the system, with Cin being the arterial blood concentration (driving concentration) and Cout (the venous blood concentration), it can easily be demonstrated that 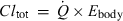 with Ebody, being the overall body extraction ratio.
with Ebody, being the overall body extraction ratio.
 ()
()It should be noted that as cardiac output is a blood flow and not a plasma flow, the interpretation of body clearance using Eqn 19 assumes that plasma clearance and blood clearance are equal (or at least not too different), i.e. the blood and plasma concentrations are equal or similar (vide infra).
The clearance model described by Eqn 17 also indicates that body clearance cannot be higher than the cardiac output (Fig. 8). For a drug exhibiting an extraction ratio equal to 1 (i.e. a total first-pass effect) for both liver and kidney, the expected Ebody is equal to about 0.5 due to the fact that the sum of liver and kidney blood flows represents about half of the cardiac output.

Maximal blood clearance in different domestic species. According to the minimal model of blood clearance, (i.e. Cl = cardiac output× extraction ratio) the maximal possible blood clearance is equal to the cardiac output when the overall extraction ratio is equal to 1. If a higher clearance is computed, attention should be paid to possible errors or misconceptions (e.g. blood concentrations much higher than plasma concentrations). If the hepatic (Eh) and renal (ER) extraction ratios are equal to 1 (i.e. if there is a first-pass effect for both liver and kidney), the body clearance should be equal to about half the cardiac output because blood flows for liver and kidney are approximately equal to 50% of the cardiac output.
If after i.v. administration (e.g. into the jugular vein), body clearance is higher or of the same order of magnitude than the cardiac output (55 mL/kg/min in horse) as reported for acepromazine (46 mL/kg/min) (Marroum et al., 1994) or pentoxyfilline (51 mL/kg/min) (Crisman et al., 1993) in the horse, consideration should be given to a possible pulmonary first-pass effect. Indeed, if the lung is able to eliminate a drug with an extraction ratio higher than 0.5, the body clearance might be greater than the cardiac output because only 50% of the administered dose will be actually available to the systemic circulation (see Chiou, 1982 for further explanation). For PgF2α in sheep, the lung extraction ratio is about 0.9, indicating that 90% of PgF2α is eliminated after a single lung passage (Bonnin et al., 1999). Other possible explanations for a very high body clearance are a direct metabolism of the drug in blood.
Figure 9 shows how to interpret body clearance for four antibiotics in dogs. This figure draws attention to the fact that body clearance and terminal half-life do not convey the same information on drug elimination. Very different terminal half-lives can be observed for drugs having the same body clearances, because terminal half-life is also influenced by drug distribution. Terminal half-life is said to be a hybrid parameter (influenced by both clearance and distribution), and it will be extensively discussed in our companion paper on terminal half-life. Thus, half-life is not an appropriate parameter to assess the intrinsic body capacity to eliminate a drug. For instance, in the horse it was shown for gentamicin in nephrotoxic conditions that terminal half-life was unchanged (192 min vs. 204 min) although plasma clearance was decreased by 40% (Riviere et al., 1983).

Interpretation of plasma clearance for four antibiotics in the dog. Typical values of plasma clearance and body overall extraction ratio for four antibiotics in the dog are reported. Penicillin, gentamicin and oxytetracycline while having the same plasma clearance also have the same overall extraction ratio (about 3–4%). In contrast, tylosin has an extraction ratio fivefold higher, indicating that the ability of the body to eliminate tylosin in the dog is fivefold higher than for the three other antibiotics. For the same antibiotics, the plasma half-lives are reported and a totally different picture appears between these antibiotics, oxytetracycline having the longest terminal half-life despite a similar clearance to penicillin and gentamicin. This is due to the fact that terminal half-life is a hybrid parameter also influenced by the extent of drug distribution, which is much larger for oxytetracycline than for penicillin.
The next step in interpreting plasma clearance involves interpreting the body extraction ratio per se by having recourse to some breakpoint values for Ebody. Figure 10 explains how to compute these breakpoints. Practically speaking, an Ebody of 0.05 or lower is generally desirable to develop a drug for oral administration with a not too high dosage regimen (vide infra). Table 1 gives the breakpoint values for high, medium and low clearances in domestic species.

Breakpoint values for high, medium and low overall body extraction ratio (E). A high, medium, or low overall body E can be calculated bearing in mind that plasma (body) clearance is given by the relation: Clbody=Clrenal+Clhepatic+Clother. The kidney and liver are the two most important contributors to drug elimination, with Clother usually negligible. For liver and kidney, E can be considered high if E > 0.70, medium if E = 0.30, and rather low if E < 0.10 (see Rowland & Tozer, 1995 for explanation). Considering that kidney (20%) and liver (30%) blood flow represent about 50% of cardiac output, overall E should be considered high if above 0.35 (0.70 × 0.50), medium if around 0.15 (0.30 × 0.50), and low if around 0.05 (0.10 × 0.50).
| Rat | Cat | Dog | Sheep | Man | Pig | Cattle/horse | |
|---|---|---|---|---|---|---|---|
| BW (kg) | 0.2 | 3.0 | 20 | 50 | 70 | 100 | 500 |
| High (E = 0.35) | 85 | 51 | 41 | 30 | 28 | 26 | 19 |
| Medium (E = 0.15) | 37 | 22 | 17.4 | 13 | 12 | 11.3 | 8.25 |
| Low (E = 0.05) | 12.2 | 7.3 | 5.8 | 6.2 | 5.8 | 5.4 | 3.6 |
- E: extraction ratio between 0 and 1.
Application of plasma clearance for computation of a dose
There are many practical uses for plasma clearance, the most relevant application being computing a dose (therapeutic, toxic…) using Eqn 1. For this, the target (therapeutic, toxic…) average plasma concentration must be known (from the literature, from a PK/PD trial, extrapolated from an in vitro assay for example for antibiotics, etc.). For phenylbutazone in the horse, therapeutic concentration was directly estimated by PK/PD modelling (3.6 μg/mL) (Toutain et al., 1994), and considering the plasma clearance (41.3 mL/kg/h), the daily dosage regimen by the i.v. route should be 3.6 mg/kg/day, which is consistent with the recommended dosage regimen. Usually, the plasma clearance is known but this is generally not the case for the plasma concentration of interest (therapeutic, toxic…); compilation of therapeutic and toxic plasma concentrations in man for several hundreds of drugs is given by Schulz and Schmoldt (1997).
For antibiotics, the drug potency is often assessed experimentally by measuring the in vitro minimum inhibitory concentration (MIC) for a given pathogen. In addition, surrogate indices to predict antibiotic efficacy have been proposed such as the area under the inhibitory curve (AUIC) for quinolones (Lees & Shojaee Aliabadi, 2002; Toutain et al., 2002).
 ()
()Equation 20 can be simplified by ignoring fu when the free fraction is dominant (e.g. for aminoglycosides) and also F for i.v. route (F = 1). Conversely, for drugs extensively bound in plasma, fu should be taken into account (Hyatt et al., 1995). For further explanation concerning Eqn 20 and its units, see Bousquet-Melou et al. (2002), Toutain et al. (2002) and Toutain (2003).
Estimation of a dose from in vitro data can be carried out for any drug for which an efficacious concentration has been obtained on the basis of an in vitro (or ex vivo) assay. For example, using a membrane feeding system, the IC99 of lufenuron (a compound for control of flea infestation in dog and cat) for Ctenocephalides felis was found to be from 50 to 100 ng/mL. Assuming a lufenuron blood clearance in the cat of about 0.56 L/kg/day, (MacKichan & Hink, 1993) Eqn 1 predicts a minimal lufenuron dose of 0.028–0.056 mg/kg/day (or 5–10 mg/kg per 6 months), which matches the experimentally determined dose of subcutaneously injected lufenuron in cats.
When estimating a dose from in vitro drug potency, it should be remembered that in vitro drug concentrations are generally free drug concentrations (Toutain & Bousquet-Melou, 2002).
The use of plasma clearance to estimate plasma therapeutic and toxic concentrations
 ()
()| Drugs | Dose (mg/kg) | Dosing interval (h) | Clearance (mL/kg/h) | EPC (ng/mL) |
|---|---|---|---|---|
| Acepromazine | 0.100 | 24 | 2600 | 1.6 |
| Bromhexine | 0.200 | 24 | 3200 | 2.6 |
| Butorphanol | 0.100 | 5 | 450 | 44 |
| Caffeine | 5.000 | 24 | 35 | 5952 |
| Carprofen | 0.700 | 24 | 8.7 | 3352 |
| Clenbuterol | 0.0008 | 12 | 350 | 0.19 |
| Codeine | 0.600 | 24 | 800 | 31 |
| Detomidine | 0.020 | 24 | 400 | 2.1 |
| Dexamethasone | 0.020 | 24 | 480 | 1.7 |
| Dipyrone | 5.000 | 24 | 300 | 694 |
| DMSO | 1000 | 24 | 80 | 520833 |
| Eltenac | 0.500 | 24 | 74 | 282 |
| Flunixin meglumine | 1.100 | 24 | 60 | 764 |
| Furosemide | 1.000 | 8 | 500 | 250 |
| Glycopyrrolate | 0.004 | 24 | 1000 | 0.17 |
| Guaiphenesin | 100 | 24 | 300 | 13889 |
| Heptaminol | 10 | 24 | 1245 | 335 |
| Hordenine | 2.000 | 24 | 4300 | 19 |
| Ibuprofen | 10 | 24 | 164 | 2541 |
| Isoxsuprine | 0.600 | 24 | 2600 | 9.6 |
| Ketamine | 2.200 | 24 | 1500 | 61 |
| Ketoprofen | 2.200 | 24 | 300 | 306 |
| Meclofenamic | 2.200 | 24 | 120 | 764 |
| Meperidine | 1.0 | 5 | 1100 | 182 |
| Methadone | 0.100 | 5 | 680 | 29 |
| Methylprednisolone | 0.400 | 24 | 1000 | 17 |
| Morphine | 0.100 | 5 | 500 | 40 |
| Naproxen | 10 | 24 | 32 | 13021 |
| Omeprazole | 0.250 | 24 | 600 | 17 |
| Pentazocine | 0.300 | 5 | 1700 | 35 |
| Phenylbutazone | 4.400 | 24 | 41.3 | 4439 |
| Quinidine | 5 | 24 | 330 | 631 |
| Salicylate | 20 | 24 | 120 | 6944 |
| Triamcinolone | 0.020 | 24 | 486 | 1.7 |
| Vedaprofen | 1.000 | 24 | 70 | 595 |
| Xylazine | 1.100 | 24 | 1200 | 38 |
The use of plasma clearance to extrapolate a dose from one species to another
 ()
() ()
() ()
()To illustrate the use of Eqn 24, the most likely dose for morphine in the dog and horse can be calculated from the dose recommended in humans, assuming equivalent drug potency between species. The recommended dose in humans is 10 mg in toto (i.e. about 0.17 mg/kg), and plasma clearance is reported to be 14.7 mL/kg/min (Stanski et al., 1978). In the dog, the morphine plasma clearance is higher (85 mL/kg/min) (Barnhart et al., 2000), which leads to an estimated dose of about 1 mg/kg for the dog. In contrast, the morphine plasma clearance in the horse is rather low (8.64 mL/kg/min) (Combie et al., 1983), which implies that a lower dose is needed in horses (0.1 mg/kg). No correction for drug binding to plasma protein is required as the extent of plasma binding is similar across all three species (Baggot & Davis, 1973).
Total clearance and dosage regimen adaptation
 ()
()By comparing the numerical value of Clhepatic and Clrenal, relevant information is obtained to predict the potential needs for dosage adjustment in animals having liver or kidney disorders. Practically speaking, if the renal clearance mainly contributes to the total clearance (e.g. more than 50% of total body clearance), a significant drug overexposure can be expected in case of renal insufficiency (e.g. renal function is <50% of physiological value), and marketing drug companies should investigate the influence of kidney function on the kinetics of such drugs in animal species prone to renal insufficiency.
Plasma clearance during early development of a new drug entity in order to document the feasibility of an oral route of administration
The plasma clearance concept can be useful in early drug development in order to address some basic questions such as: can the drug be successfully administered as an oral dosage form or conversely, will the drug undergo an extensive hepatic first-pass effect leading to a low and variable systemic bioavailability?
The question can be answered from a single i.v. trial by measuring both total and renal clearance and thus allowing the evaluation by difference of a non-renal clearance, which in the first instance can be attributed to liver clearance.
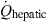 ) and the hepatic extraction ratio (Ehepatic), i.e.
) and the hepatic extraction ratio (Ehepatic), i.e.
 ()
() ()
() ()
()As  is known (about 30% of the cardiac output), the Fmax can be assessed easily by evaluating only Clhepatic, which in turn requires knowledge of both Cltot and Clrenal (Eqn 15). The computed Fmax is the real oral bioavailability if the drug is totally absorbed by the digestive tract, i.e. if the loss of drug is only due to the hepatic first-pass effect. Using this approach, the low oral bioavailability of some drugs can easily be predicted. For example, in the dog, propranolol is totally absorbed after oral administration, and is completely eliminated by a high hepatic clearance (41 mL/kg/min). This explains the relatively low measured bioavailability despite total absorption (16 ± 12%) (Bai et al., 1985). Such a low systemic bioavailability was expected because the hepatic clearance of propranolol is of the same order of magnitude as hepatic blood flow.
is known (about 30% of the cardiac output), the Fmax can be assessed easily by evaluating only Clhepatic, which in turn requires knowledge of both Cltot and Clrenal (Eqn 15). The computed Fmax is the real oral bioavailability if the drug is totally absorbed by the digestive tract, i.e. if the loss of drug is only due to the hepatic first-pass effect. Using this approach, the low oral bioavailability of some drugs can easily be predicted. For example, in the dog, propranolol is totally absorbed after oral administration, and is completely eliminated by a high hepatic clearance (41 mL/kg/min). This explains the relatively low measured bioavailability despite total absorption (16 ± 12%) (Bai et al., 1985). Such a low systemic bioavailability was expected because the hepatic clearance of propranolol is of the same order of magnitude as hepatic blood flow.
Blood vs. plasma clearance
 ()
() ()
()Considering a haematocrit value of 0.4, Clblood is equal to about 1.66 × Clplasma, so that plasma clearance underestimates blood clearance. Therefore, for a drug almost wholly excluded from red blood cells, the maximum possible error when using plasma clearance as a surrogate of blood clearance is no greater than 40%.
In contrast, when a drug accumulates in RBC (e.g. pimobendan, chloroquine, cyclosporin, acetazolamide…), Clplasma may considerably overestimate Clblood, making the interpretation of plasma clearance incorrect in terms of blood flow. That is why knowledge of the B/P ratio is required very early in drug development, as this will determine whether the appropriate matrix for PK studies is blood or plasma.
The B/P ratio varies between a value of (1 − H) for drugs highly bound to plasma protein or/and poorly partitioned between RBC and plasma, to a possibly large value when drug has a high affinity for RBC (e.g. affinity for haemoglobin…). When B/P is higher than 1, it can be useful to know if the partitioning/departitioning of the drug between plasma and RBC is rapid or not with regard to the transit time through the clearing organ (liver and kidney). Indeed, the presence of drug in RBC is of relevance for PK interpretation only if the exchange between RBC and plasma is sufficiently rapid to participate in the drug clearance process. In other words, if during blood transit throughout the liver (about 10 sec), the kidney cortex (about 2 sec) or the renal medulla (about 30 sec), a drug located in RBC (e.g. the fraction bound to the RBC membrane) can immediately return to plasma to re-equilibrate with plasma which is stripped of the drug during the transit time, the blood concentration and blood clearance are relevant. In contrast, if the departitioning is so sufficiently slow that drug removal from the plasma does not lead to significant re-equilibration during organ transit, plasma concentration becomes the relevant concentration, because then, RBC should be regarded as a peripheral (circulating) tissue compartment and not a central one. This is the case for para-aminohippuric acid (PAH), which is used to assess kidney circulation. PAH undergoes a first-pass effect during its passage throughout the kidney, but only PAH located in plasma is involved. Thus, PAH plasma clearance measures an effective plasma flow and not a blood flow. For further explanation see Hinderling (1997) and Wilkinson (1987).
Arterial versus venous blood sampling: the question of the jugular vein
In veterinary medicine, blood administration and sampling are often carried out at the venous level, which can lead to two possible drawbacks. The first is related to drug administration and to a possible pulmonary first-pass effect, which can reduce the available (systemic) dose by a factor equal to the pulmonary extraction ratio (vide supra). The second difficulty is related to blood sampling from venous vessels and the possibility that the venous plasma concentration may differ from that of arterial plasma. Indeed, for clearance computation, plasma concentration measured at the venous level is assumed to be a surrogate of the simultaneous plasma concentration at the arterial level. This is true only if there is no significant metabolic site between the aorta and the sampling site. This assumption is reasonable when the blood is sampled on the arm in man because the venous blood has only crossed muscles, but it is more debatable for jugular blood which is more often sampled in animals as the blood has crossed the entire head (brain, possible saliva elimination in ruminants, etc.)
Conclusion
Total body clearance is the most important PK parameter and needs to be evaluated in vivo for any drug through an i.v. study. The main use of clearance is to compute or adapt a dosing rate. When interpreted in physiological terms, it provides an understanding of some basic features of drug disposition.

