

Transcriptome response of the Pacific oyster (Crassostrea gigas) to infection with Vibrio tubiashii using cDNA AFLP differential display
Summary
We used qualitative complementary DNA-amplified fragment length polymorphism (cDNA-AFLP) differential display analysis and real-time, quantitative PCR (RT-qPCR) to identify genes in the Pacific oyster Crassostrea gigas, whose transcription either changes in response to exposure to a pathogenic bacterium (Vibrio tubiashii) or varies between families known to differ in sensitivity to heat stress, before and at 12 and 36 h after bacterial exposure at a temperature of 25 °C. These conditions simulate those associated with summer mortality syndrome, a poorly understood cause of massive mortalities in cultured Pacific oysters in North America, Asia and Europe. Using 32 AFLP primer pairs, we identified 92 transcript-derived fragments that are qualitatively differentially expressed. We then cloned and sequenced 14 of these fragments, designed fragment-specific primers and quantified their transcription patterns using RT-qPCR. Most of the differences in transcription patterns between stress-tolerant and stress-sensitive families were evident before bacterial exposure, and genes that responded to bacterial exposure did so in parallel between stress-sensitive and stress-tolerant families. blast searches of sequence databases revealed that these fragments represent genes involved in immune response as well as genes related to metabolic processes. Our data support the hypothesis that family level differences in resistance to stress in Pacific oysters are largely attributable to constitutive differences in gene transcription or ‘general vigour’ that are detectable before and maintained after infection, rather than being due to induced responses at the transcriptome level.
Introduction
Summer mortality syndrome is a widespread but poorly understood phenomenon affecting Pacific oysters, Crassostrea gigas. It was identified as early as the 1970s in Japan (Koganezawa 1975) and has since been reported in the United States (Glude 1975), France (Maurer & Comps 1986) and Mexico (Chávez-Villalba et al. 2007). In the Pacific Northwest region of the USA, summer mortality results in massive die-offs of near-market-size oysters, making it particularly costly for growers. To date, no single pathogen or environmental factor has been identified as the sole cause of summer mortality. Rather, evidence is accumulating that mortalities result from opportunistic pathogens that infect oysters, already weakened by a combination of stress and the high energetic costs associated with reproduction, because summer mortality most frequently occurs during periods of prolonged summer water temperatures greater than 19 °C in eutrophic bays or estuaries in which oysters both grow rapidly and also invest heavily in reproduction (Ventilla 1984; Moal et al. 2003; Soletchnik et al. 2003; Samain & McCombie 2008). Temperature stress and other environmental factors (Perdue et al. 1981; Cheney et al. 2000) thus appear to compromise the oyster’s resistance to a variety of opportunistic pathogens (Burge et al. 2003), and while pathogens are the proximate cause of summer mortality, the deeper causes are likely to be rooted in the more general stress response of oysters rather than in pathogen-specific induced responses.
During the last decade, researchers have devoted considerable effort to understand how Pacific oysters respond to both biotic and abiotic stress, with the goal of reducing losses due to summer mortality. Most interestingly, selection experiments conducted in France, where summer mortality is very predictable, have provided convincing quantitative genetic evidence that resistance to summer mortality is highly heritable (Dégremont et al. 2005). However, although candidate genes have been proposed to be involved in physiological pathways affecting susceptibility to summer mortality (Huvet et al. 2004; David et al. 2007), the specific genes controlling resistance have not been clearly identified. Selective breeding targeting resistance to summer mortality in the USA is, however, more complicated because, in contrast with the situation in France, summer mortality in the USA is temporally sporadic and spatially variable. This, combined with the lack of a single causative pathogen, makes it difficult to identify appropriate sites for field trials or even to measure the trait of interest (survival under summer mortality conditions), reliably and consistently. When this is combined with our ignorance about the specific genes involved in stress tolerance, it makes developing improved lines with resistance to summer mortality difficult, because shellfish breeders in the USA cannot rely upon consistent challenges and selection pressure in the field, cannot impose appropriate challenges in the laboratory and cannot directly target genetic-level differences using marker-assisted selection.
In this study, our objective was to develop a better understanding of the genetic-level responses of oysters to both temperature stress and pathogenic bacteria, and to identify specific genes that contribute to survival under stressful conditions by comparing the genetic transcription profiles of experimentally stressed and control oysters, from full-sib families known to differ in stress tolerance. Our motivation is that a genome-wide examination of differences in the patterns of gene transcription in response to known stressors and comparisons between families with known differences in stress tolerance will likely identify major genes that contribute to stress tolerance, and also that these genes could be useful in future studies aimed at developing a programme of marker-assisted selection in conjunction with quantitative trait locus mapping strategies (e.g. Sauvage 2008). Immune-related genes have already been reported in oyster haemocytes based on expressed sequence tags (ESTs) using a targeted gene approach (Montagnani et al. 2001, 2004, 2007; Gueguen et al. 2003; Badariotti et al. 2007; Gonzalez et al. 2007; Lelong et al. 2007).
Pacific oysters are a non-model organism, and only limited sequence information is currently available, although this situation is rapidly improving (Tanguy et al. 2008). The only currently available cDNA microarray, for example, contains only a relatively small fraction of the Pacific and Eastern oyster genomes (Jenny et al. 2007). While we are also employing this technology to study the stress responses of Pacific oysters, in order to access a larger proportion of the transcriptome in this study, we used a differential display-type method, complementary DNA-amplified fragment length polymorphism (cDNA-AFLP). cDNA-AFLP (Bachem et al. 1996) uses restriction enzyme digestions of cDNA libraries derived from ESTs followed by selective amplification of a subset of the digestion products based on techniques originally developed by Vos et al. (1995) for genomic DNA. cDNA-AFLP is a mature, fast and reproducible method of surveying gene transcription patterns and can unravel the molecular basis of biological systems without the need for extensive sequence data.
In this study, we used cDNA-AFLP to characterize transcriptome-level changes in gene expression in haemocytes of Pacific oysters before and at 12 and 36 h following bacterial challenges with Vibrio tubiashii at a temperature of 25 °C, conditions which in nature are associated with summer mortality. We also compared the responses of families known to differ in their sensitivity to heat shock, which is both an important contributing factor in summer mortality and has also been demonstrated to have significant impacts on cell-mediated immunity in C. gigas (Li et al. 2007).
Methods
Biological materials
To identify oyster families that differed in their stress tolerance, in the fall of 2005, we heat-shocked (39 °C, 1 h) one hundred juvenile oysters (5 months old, 1–2 cm) from each of 44 families from a cohort of full-sib oyster families produced according to Langdon et al. (2003), as part of an ongoing selective breeding programme. Based on their survival at 6 d after this stress, we classified four families as low-surviving (L) or high-surviving (H) if their survival after heat shock as juveniles was <30% or >70% respectively. Several hundred unstressed oysters from each of these L and H families were held in flow-through troughs supplied with sand-filtered seawater from Yaquina Bay estuary, Newport, Oregon at ambient temperature for approximately 6 months. During the summer of 2006, we transferred the animals from troughs to experimental trays (described below) and gradually acclimated them from 12 °C to the experimental temperature of 25 °C over a period of 6 days. This was carried out by raising the temperature 2 °C per day to avoid triggering acute heat stress responses in the oysters. Once the temperature in the trays reached 25 °C, oysters were held at this temperature for 2 days before exposure to bacteria.
The bacterial strain used in this study (ATCC 19106) was kindly supplied by Ralph Elston (AquaTechnics/Pacific Shellfish Institute). The strain was originally collected from Pacific oyster larvae and juveniles (Estes et al. 2004).
Experimental design
Each replicate experimental unit consisted of 12 oysters from each of the four families (two H families, two L families for a total of 48 oysters) held in 8-l flow-through trays, and the full experimental set-up consisted of four such trays. We sacrificed four oysters from each family and collected haemolymph samples at three time-points: (i) prior to bacterial challenge (T0 = control), (ii) 12 h after challenge (T12 h) and (iii) 36 h after challenge (T36 h). At each sampling time, we removed the upper valve from the sampled oysters, collected haemolymph from the pericardial cavity using a 1 ml syringe fitted with a 20 G needle, and pooled the four samples from individual oysters within a family to produce a single haemolymph pool for each family from each replicate at each sampling time. The volume of these pools varied, but generally ranged from 0.5 to 1 ml. When collecting these samples, we emptied the syringe directly into sample-specific 15 ml conical centrifuge tubes containing 3 ml of chilled RLT lysis buffer (Qiagen), and when all four individual samples had been pooled, disrupted the cells by repeated aspiration using the same syringe and needle. Samples were then frozen at −80 °C for later analysis.
After removing the unchallenged oysters from the experimental trays, we subjected the remaining eight oysters/family/tray to a bacterial challenge by turning off the incoming water flow and adding log-phase V. tubiashii culture (ATCC 19106) (Tubiash et al. 1965; 1970) to the trays for 3 h in static water, after which we restored water flow and allowed the bacteria to flush gradually from the trays. Prior to this exposure, we grew bacteria at 25 °C for 36 h in Marine Broth culture medium (Difco), used a barium sulphate turbidity standard to estimate the bacterial density, and then calculated the volume of bacterial suspension required to attain an initial ambient concentration of 106 colony forming units (CFU) per ml in the trays at the time of inoculation.
Bacterial count in oyster tissues
At each of the three sampling times, we pooled and weighed the remaining tissues from the sampled oysters by family type (i.e. high- and low-survival) and experimental replicate, and ground the tissue pools to a slurry using a Polytron homogenizer (Kinematica). We then quantified the bacterial concentration in these oyster tissue pools by serially diluting these homogenates in phosphate-buffered saline, filtering on a 0.45-μm pore size membrane, plating on Vibrio selective agar (Thiosulphate Citrate Bile Salts Sucrose Agar; TCBS, Difco) plates and incubating overnight before counting colonies. We expressed these counts as colony forming units per gram of oyster tissue (CFU/g) for statistical analysis.
RNA extraction and amplification
We extracted total RNA from each haemolymph pool using the QIAGEN RNeasy Mini Kit following the manufacturer’s protocol. In vitro RNA amplification was used to increase the amount of material available for analysis by using the SenseAMP Plus RNA Amplification Kit (Genisphere), according to the manufacturer’s instructions. After treating the amplified samples with DNAse I (Qiagen, RNase-Free DNase Set), 500 ng samples of poly-A senseAMP RNA were reverse-transcribed using SuperScript™ III Reverse Transcriptase (Invitrogen) following the manufacturer’s protocol for oligo dT priming. Second strand synthesis was performed using DNA Polymerase I (Escherichia coli) and RNAse H (New England Biolabs) in a total volume of 100 μl.
Qualitative cDNA-AFLP analysis
Before cDNA-AFLP analysis, we first quantified the cDNA concentrations of each family and replicate-specific sample using a Nanodrop spectrophotometer and combined appropriate volumes from the two H and two L families from each of the four replicate trays (eight samples total) to ensure equal representation among families in the pooled samples at each of the three sampling times. This pooling produced a total of six cDNA template pools (H and L families at T0, T12 h and T36 h).
We then digested 50 ng of the cDNA templates obtained above with two restriction enzymes, EcoRI and MseI (New England Biolabs) and simultaneously ligated the resulting fragments to double-stranded adapters specific to the EcoRI and MseI ‘sticky ends’ in the same reaction using T4 Ligase (New England Biolabs). The adapters used were: EcoRI: 5′-CTC GTA GAC TGC GTA CC-3′ and 5′-AAT TGG TAC GCA GTC TAC-3′; MseI: 5′-GAC GAT GAG TCC TGA G-3′ and 5′-TAC TCA GGA CTC AT-3′. This digestion–ligation step was performed in a total volume of 50 μl and incubated at 37 °C for 3 h. We performed non-specific PCR amplifications using primers with one additional nucleotide at the 3′ ends of the enzyme-specific adapters using 20 cycles of 94 °C for 30 s, 56 °C for 1 min, 72 °C for 1 min with a 5-μl aliquot of a 1:10 dilution (in a 0.1× Tris EDTA buffer) of the digestion ligation product as template in a reaction volume of 50 μl.
Following this first non-specific amplification, we diluted the PCR products 10-fold and used 5 μl aliquots as template for selective amplification with 32 EcoRI +2 and MseI +2 primer combinations (Table S1). We precipitated the amplification products by adding 10% 3 m sodium acetate and 2 volumes of absolute ethanol and freezing overnight at −20 °C. For each selective primer combination, 3 μl of each of the PCR products from each of the six pooled samples was separated into measurable fragments using electrophoresis through 6% polyacrylamide gels run at 1000 V for 2 h using a model S2 sequencing gel electrophoresis apparatus (Gibco BRL). The resulting cDNA AFLP fragments were visualized by silver staining according to the procedure of Bassam et al. (1991) using a 100-bp DNA ladder (1 μl) (Promega) as a size standard.
Isolation, cloning and sequencing of re-amplified DNA fragments.
After staining, we excised 92 bands of interest from the gels (see Results for details) and eluted them in 50 μl of sterile distilled water at 4 °C for about 16 h and re-amplified them using the same EcoRI +2/MseI +2 primer combination and selective PCR conditions that produced them. The re-amplified fragments were visualized in 2 % agarose gels, and bands were excised and purified using the QIAquick Gel extraction Kit (Qiagen). This procedure was repeated until the band of interest was purified.
An aliquot of the re-amplified DNA was then cloned into a pCR4-TOPO vector using the TOPO TA cloning for sequencing kit (Invitrogen). Plasmid DNA was purified from 7 ml of an overnight culture of E. coli in LB medium using the Wizard®Plus SV Miniprep DNA Purification System (Promega), and sequenced using the Big Dye Terminator v.3.1 Cycle Sequencing kit on an ABI 3730 XL DNA sequencer (Applied Biosystems). We used BLASTX (Altschul et al. 1997) to ascertain similarity of these 92 fragments to known proteins, resulting in 33 matches (see Results for details).
Real-time PCR
Real-time quantitative PCR (RT-qPCR) analyses did not use the doubly pooled samples that were used for qualitative cDNA-AFLP, but rather used the remaining poly-A senseAMP RNA from the family, time and tray-specific pools to exploit the experiment’s replicated design. From these replicated samples, we synthesized first strand cDNA using 500 ng of DNA-free poly-A sense AMP RNA, following the instructions of the high capacity cDNA archive kit (Applied Biosystems), and for each of the fragments chosen for verification, we designed primers (Table S2) based on our sequencing results using primer express version 2.0.0 (Applied Biosystems). We used Elongation factor 1α (GenBank: AB122066; F: GGAAGCTGCTGAGATGGGAA, R: TCCAACACCCAGGCGTATTT) as reference housekeeping gene in our RT-qPCR analyses (coefficient of variation <5% across samples; Huvet et al. 2004; Fabioux et al. 2004; Labreuche et al. 2006; Fleury et al. 2008; Gagnaire et al. 2007; Taris et al. 2008). We tested a minimum of seven primer pairs for each gene to avoid artefacts caused by sequence polymorphism on the final outcome (Taris et al. 2008). For each RT-qPCR assay, we used 5 ng of cDNA as template in a 25-μl reaction that included 50 nm (final concentration) of each primer and SYBR Green PCR master mix (Applied Biosystems). PCR cycling conditions were: 50 °C for 2 min (AmpErase® UNG activation), 95 °C for 10 min (AmpliTaq Gold® DNA polymerase activation), 50 cycles of 95 °C for 15 s and 60 °C for 1 min and finally 95 °C for 15 min, 60 °C for 15 s. RT-qPCR reactions were run and results were analysed using an Applied Biosystems 7500 Real Time PCR system (software version 1.4). We expressed our data as concentrations relative to the Elongation factor 1α reference gene by normalizing raw CT values using target/reference ratios [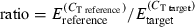 ], where E represents the empirically determined efficiency estimated for each reaction using linregpcr software (Ramakers et al. 2003).
], where E represents the empirically determined efficiency estimated for each reaction using linregpcr software (Ramakers et al. 2003).
Statistical analysis
Bacterial count in oyster tissues

Real-time PCR

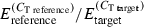 ), μ is the overall mean, αi is the family type effect, βj is the family effect nested with family type, γk is the sampling time effect, δl the replicate effect, intik is the interaction between family type and time and εijkl the residual error. As for the previous analysis, we performed planned comparisons using linear contrasts as implemented by the contrast statement in the glm procedure. A priori contrasts were included to test for significant differences between family types at each of the three time points and also for time effects within each family type. The consistency of transcription of the reference gene was tested by using the same model with the exception of the dependent variable (
), μ is the overall mean, αi is the family type effect, βj is the family effect nested with family type, γk is the sampling time effect, δl the replicate effect, intik is the interaction between family type and time and εijkl the residual error. As for the previous analysis, we performed planned comparisons using linear contrasts as implemented by the contrast statement in the glm procedure. A priori contrasts were included to test for significant differences between family types at each of the three time points and also for time effects within each family type. The consistency of transcription of the reference gene was tested by using the same model with the exception of the dependent variable ( ).
).Results
Bacterial counts
Before the V. tubiashii inoculation, the concentration of bacteria able to grow on TCBS media did not exceed 3.1 × 101 CFU/g. In contrast, the bacterial concentration in oyster tissues was significantly higher than this pre-inoculation level at both 12 h (χ2 = 246.5, P < 0.001) and 36 h post-inoculation (χ2 = 54.5, P < 0.001) (Table S3). Furthermore, bacterial concentrations tended to decrease between these two sampling times, with all the values exceeding 103 and reaching a maximum of 2.2 × 104 CFU/g at 12 h and ranging from 2.7 × 101 to 5.9 × 102 CFU/g at 36 h (Fig. 1, Table S3). No significant effects of family type, family within-type or replicate were detected within any of the sampling times.

Bacterial count within oysters (colony forming unit/g) per family type [two families for each type designation, namely high-surviving (H) and low-surviving (L) group], experimental replicate and date (pre-inoculation = control; 12 h and 36 h post-inoculation).
Gene transcription analysis: cDNA-AFLPs profiles
We tested 32 combinations of AFLP primers that produced slightly more than 800 transcript-derived fragments (TDF) resulting in an average of approximately 25 distinct AFLP bands per primer pair. Ninety-two TDFs displaying reliable band intensity were qualitatively identified as differentially expressed, ranging in size from 94 to 391 bp.
Similarity with known genes
Table 1 lists a subset of 33 clones that matched known genes in GenBank with an E-value threshold of less than 10−1, along with a qualitative representation of their pattern of differential transcription among family types and sampling times. We used a liberal threshold so as not to exclude potentially interesting genes such C1q (see Table 1). Table 2 reports the qualitative transcription patterns of the other 59 TDFs with either no Genbank homology or homology with E-values >10−1. The 33 clones with known homology fell into eight groups based on gene functions: (i) cell/organism defence, (ii) detoxification/stress protein, (iii) cell cycle, (iv) transcriptional regulation, (v) cell signalling, (vi) metabolism, (vii) ribosomal protein and (viii) unknown function.



Qualitative cDNA-AFLP banding patterns
As shown in Tables 1 and 2, differential transcription was observed in a wide range of patterns among sampling times and family types. However, most of the profiles consisted of constitutive differential transcription among family types throughout the time–course series, which is indicated by alternating patterns of dark and light bands. That is, the L and H family types differed in their transcription levels for most of these genes even before bacterial exposure, and these differences were either maintained after the bacterial challenge or show relatively minor variation in their response to the challenge.
Quantitative-PCR
Following the qualitative cDNA-AFLP analysis, we used RT-qPCR to evaluate quantitatively and confirm the transcription patterns of 14 selected fragments. As we were only able to extract a limited quantity of RNA from small haemocyte samples, we had limited quantities of cDNA available for RT-qPCR and were thus able to examine quantitatively only a subset of the genes that showed differential transcription using cDNA-AFLP. We made these choices based upon the gene functions listed in GenBank and their potential relationship to pathogenic bacteria, and selected fragments that BLASTX searches indicated were found to encode homologues of the following 14 proteins. Each of these has been assigned a letter (listed in Table 2) in addition to the GenBank accession number to which it matched to simplify references to figures : (A) C1q domain protein (E-value = 9E-01; FC325715), (B) C-type lectin 2 like protein (E-value = 4E-34; EX956425), (C) legumain precursor (E-value = 3E-11; EX956424), (D) cavortin (E-value = 9E-22; EX956449), (E/F) cytochrome P450-like (E-value = 3E-10; EX956386/E-value = 4E-6; EX956383), (G) pyrroline-5-carboxylase synthase (E-value = 3E-11; FD483994), (H) Rho GTPase (E-value = 6E-5; FD483988), (I) d-lactate dehydrogenase (E-value = 3E-01; EX956442), (J) tyrosine phosphatase-related protein (E-value = 2E-05; EX956370), (K) putative Universal stress protein Usp (E-value = 8E-14; EX956445), (L) prostaglandin E receptor 4 (E-value = 3E-03; EX956398), (M) transglutaminase (E-value = 9E-02; FE192420) and (N) similar to S-crystallin (E-value = 3E-05; EX956399). The transcription profiles obtained by RTq-PCR are summarized in Fig. 2, along with the observed cDNA-AFLP pattern. Statistical outcomes are shown in Table S4.

Quantitative transcription of 14 selected fragments relative to Elongation factor 1α. Comparison was made between the two groups of oysters, namely high-surviving (H, black bars) and low-surviving (L, white bars) in response to acute heat shock as juveniles. The three time-points are represented from each fragment (control = prior to inoculation; 12 h and 36 h post-inoculation). Each bar represents four replicates of two families (±SE). Below figure the corresponding AFLP profiles.
For most of the genes tested, RT-qPCR results followed the same transcription profile as cDNA-AFLP (Fig. 2), with the notable exception of transcripts H (Rho GTPase) and M (transglutaminase). Addressing these exceptions first, for fragment H (Rho GTPase), the RT-qPCR data indicate that the H families have higher levels of transcription (F = 6.76, P < 0.05), while cDNA-AFLP profiles suggested the opposite tendency. The RT-qPCR result of transcript M (transglutaminase) displayed no significant differences in transcription across family types for the three time points (general model, F = 0.07, P = 0.80), although its cDNA-AFLP profile seemed to indicate qualitative differences in gene transcription between family types at T0 and T12.
Turning to the other transcripts, aside from one fragment J (tyrosine phosphatase related protein), we found no significant family type-by-time interactions, in agreement with the constitutive up- or down-regulation patterns observed with the cDNA-AFLP profiles. Of the 12 remaining transcripts, six displayed a significant family type effect in the general model [A (C1q domain protein), E and F (cytochrome P450-like), I (d-lactate dehydrogenase), K (putative Universal stress protein Usp), and L (prostaglandin E receptor 4)] with A (C1q domain protein) and L (prostaglandin E receptor 4) consisting of higher transcription levels in L families compared with H families over the whole time series. A similar pattern was detected for transcripts I (d-lactate dehydrogenase) and K (putative Universal stress protein Usp), but the family type effect was significant at only two time points. For fragments E and F (cytochrome P450-like protein), the RT-qPCR assays indicated higher transcription in H families after bacterial challenge, but the two profiles had different temporal patterns. For the other transcripts, significant effects of families nested within family types seemed to ‘dilute’ the family type effect in the form of among-family variation despite similar family classification. This appeared to be particularly true for B (C-type lectin 2 like protein), D (cavortin), and to a lesser extent for N (similar to S-crystallin), G (pyrroline-5-carboxylase synthase) and C (legumain precursor). Finally, Transcript J (tyrosine phosphatase related protein) showed a significant family type effect (up-regulation in H lines), but only 12 h post-challenge.
Accounting more specifically for the time component in the general model, we found that the gene transcription of seven transcripts (C, E, G, H, J, K, N) varied significantly among sampling times following two opposite trends: (i) a decrease in gene transcription 12 h post-challenge followed by an increase 24 h later [C (legumain precursor); K (putative Universal stress protein Usp); E (cytochrome P450-like) and A (C1q domain protein), although the time effect is not significant (F = 2.57, P = 0.09)]; (ii) an increase in transcription 12 h after infection followed by a decrease 24 h later [J (tyrosine phosphatase-related protein); G (pyrroline-5-carboxylase synthase); N (similar to S-crystallin); H (Rho GTPase)].
Discussion
Studies of the genome-wide transcriptional changes can elucidate the gene regulatory network(s) underlying organismal-level responses to environmental stressors, and thus serve as the first step toward identifying candidate genes for marker-assisted selection. In this study, we used a combination of qualitative cDNA-AFLP analysis and RT-qPCR to obtain further insights into the transcriptome-level responses of the Pacific oyster to two of the causes of summer mortality syndrome in the Pacific Northwest region of the USA, heat stress and bacterial infection. To accomplish this, we characterized gene transcription after bacterial challenge with V. tubiashii at a temperature of 25 °C, a scenario that approximates environmental conditions associated with summer mortality. In this context, cDNA-AFLP is a comprehensive method, applicable for comparative gene transcription in a variety of biological contexts for a wide range of organisms (Vuylsteke et al. 2007) and is particularly attractive because no prior knowledge of sequences is required.
Using this approach, we identified 92 transcripts as differentially expressed either between L and H families or in response to bacterial exposure. Sequence analysis and BLASTX searches of these sequences identified 33 fragments that match known genes (E < 10−1) in the GenBank database. These genes fall into eight functional classes, which we discuss in detail below. Interestingly, we found that the patterns of differential transcription between family types are mostly constitutive with regard to bacterial exposure and in that the observed differences in transcription between stress-tolerant and stress-sensitive families were present before we challenged them, and those genes whose transcription levels did change in response to bacterial exposure did so in parallel in both family types. Only one fragment J (tyrosine phosphatase-related protein) showed a significant family type*time interaction indicating differences in the time–course of induction.
There were two cases [fragments H (Rho-GTPase) and M (transglutaminase)], where AFLP results did not match the results of quantitative PCR, and these discrepancies must be addressed. One potential source of error in cDNA-AFLP is the possibility that multiple transcripts of the same size are visualized as a single band, resulting in misleading results for the transcription, for the presumably sequenced fragment (Cappelli et al. 2005). In addition, insertion/deletion mutations between restriction sites or polymorphisms within restriction sites that prevent enzymatic digestion may produce artefactual differences in the apparent transcription levels of transcript-derived fragments (Tate et al. 2006). This is particularly true for oysters, which are rather notorious for having high levels of sequence polymorphism (Hedgecock et al. 2004; Sauvage et al. 2007). Finally, polymorphism issues may impact RT-qPCR assays if they occur in priming regions, and thus affect the efficacy or efficiency of PCR (Taris et al. 2008).
Cell/organism defence
Of the fragments annotated in GenBank related to cell defence, four showed higher levels of transcription in the L oyster families than in H ones, either in a constitutive manner before the bacterial challenge (C1q and prostaglandin receptor) or as an induced response to infection (C-type lectin protein cavortin). C-type lectins function as non-self pattern recognition molecules and participate in responses to infection through a multivalent carbohydrate-binding capability (McGreal et al. 2004; Yamaura et al. 2008). Cavortin is a major haemolymph protein that includes a superoxide dismutase-related domain (Gonzalez et al. 2005), indicating that this gene is possibly associated with cell defence against reactive oxygen species (ROS) produced by oxidative stress (Huvet et al.2004). Recently, ROS production has been documented as a major element differentiating summer mortality susceptible and resistant oyster strains in France (Delaporte et al. 2007; Lambert et al. 2007). These studies found that the ROS production was significantly higher in susceptible oysters than in resistant oysters.
In addition, transcription levels of the genes C1q and prostaglandin receptor E4 were constitutively higher in L families. C1q has been previously identified in two oyster EST libraries (Jenny et al. 2002; Rafferty & Powell 2002), but these authors provide no further information about its biological function in oysters. In other organisms, the C1q domain-containing proteins (C1qDC) include a wide range of signalling molecules that are known to participate in the control of inflammation, adaptive immunity and energy homeostasis (Kishore & Reid 2000). C1qDC proteins have been described in numerous higher vertebrates as related to adaptive immunity via complement activation and have been shown to play a role in innate immunity by Yuste et al. (2006) via mechanisms that are apparently independent of complement activation. It is therefore interesting to find this protein in invertebrate species such as the sea urchin and even in eubacterial species (Tom Tang et al.2005), and to observe family level differences in its transcription.
Prostaglandins are oxygenated compounds derived from polyunsaturated fatty acids that play various physiological roles including mounting immune and inflammatory responses (Rowley et al. 2005). In molluscs, prostaglandin production has been linked with chemical defence (Di Marzo et al. 1991), maintaining ionic balance in the gills (Freas & Grollman 1980; Saintsing et al. 1983) and inducing spawning (Osada et al. 1989). The impacts of specific prostaglandins, however, are determined by the array of receptors expressed in cell membranes and the intracellular pathways to which they are coupled.
Transglutaminase appeared to be constitutively more highly expressed in H families according to our cDNA-AFLP profiles, but this was discordant with the RT-qPCR results. Transglutaminases are a family of enzymes that catalyse the formation of a covalent bond between a free amine group (e.g. protein- or peptide-bound lysine) and the gamma-carboxamid group of protein- or peptide-bound glutamine. Bonds formed by transglutaminase result in cross-linking and polymerization of proteins (Folk & Finlayson 1977). Recently, a full length cDNA that encodes a transglutaminase has been characterized in Chinese shrimp (Fenneropenaeus chinensis) and is associated with bacterial challenge (Liu et al. 2007a). It is possible that comparatively higher levels of constitutive transglutaminase transcription and translation may contribute towards enhanced disease resistance in bivalve molluscs.
Like transglutaminase, fragment C (legumain) is more highly expressed in H families even before bacterial exposure. Legumain is an asparaginyl endopeptidase that belongs to the cysteine peptidase family, known to show strict specificity for hydrolysis of asparaginyl bonds (Chen et al. 1997). Reported from diverse sources such as plant, invertebrate parasites and mammals, legumain is described as having a wide range of functions. In mammals, legumain-like cysteine peptidases have been described in antigen presentation (Maehr et al. 2005) and might have an important role in extracellular matrix remodelling (Morita et al. 2007), but its exact role remains poorly understood. Further investigation is needed to confirm the functional nature of this gene in the Pacific oyster in the context of stress resistance.
Taken together, we observed higher transcription levels, before and after bacterial exposure, of immune-relevant genes in the L families. Although more work is needed to clarify the functions of the protein products of these genes, our data contribute to a trend in which L oyster families transcribe otherwise beneficial genes at higher levels, which may either reflect a general sensitivity to stress or may lead to sensitivity to stress (Hawkins et al. 1989; Lang 2008). For example, Lang (2008) found that L families transcribed a number of otherwise beneficial genes before and after experimental heat shock, including one coding for galectin, which is known to promote phagocytosis of phytoplankton and pathogenic bacteria (Tasumi & Vasta 2007), and a gene for peroxinectin, which also encourages phagocytosis of pathogens by haemocytes in crustaceans (Liu et al. 2007b). The higher pre-stress transcription of C1Q and prostaglandin E receptor 4, and the higher induced transcription of C-type lectin protein and cavortin in L families, suggest that resistance to pathogenic infection is not necessarily achieved by increased activation of inducible defence mechanisms in response to pathogen exposure. Oysters are in constant contact with pathogenic bacteria as they filter water. One interpretation of our data is that instead of higher protein production resulting in greater survival, greater protein production is needed by these families to resist otherwise persistent attacks by pathogens.
Detoxification/stress
Reactive oxygen species and xenobiotics pose a continuous threat to sessile marine bivalves such as the Pacific oyster. Cytochrome P450 is a diverse superfamily of haemoproteins that occupies a central role in oxidative metabolism in bacteria, plants and animals (Werck-Reichhart & Feyereisen 2000), and in this study, L families transcribed CYP450 at lower levels before and after exposure to V. tubiashii. Similarly, RT-qPCR results tend to indicate a relatively lower level of transcription of gene S-crystallin in L families, despite a non-significant family type effect. Looking at the conserved domain displayed by the sequence of this fragment, we interestingly found homology with the glutathione S-transferase family of enzymes (GST; Class Sigma-like, E-value = 5E-09). This is similar to the findings of Lang (2008) who found that GST transcription was greater in whole spat of H families after heat shock. GSTs are important detoxification enzymes that catalyse the conjugation of glutathione (GSH) with a wide range of endogenous and xenobiotic agents, including environmental toxins and the products of oxidative stress (Salinas & Wong 1999). It is unclear whether this fragment truly encodes crystallin or GST, and further characterization of the fragment is needed. Interestingly, Lang (2008) found that transcription of an EST similar to S-crystallin was greater in gills of L families after heat shock, and thus it is possible that while in specific tissues a greater transcriptional response will occur in L families, at the whole body level, those systems become deficient. Nonetheless, we speculate that L families may be more prone to damage by oxidative stress and toxic organic compounds that result from downstream reactions of ROS with lipids and other molecules (Storey 1996). This is consistent with Samain et al. (2007), who found that oyster families that were susceptible to summer mortality in France had lower enzyme activity in gill of the ROS detoxification enzyme catalase (Samain et al. 2007). Lower enzyme activity could result in higher ROS availability, resulting in greater concentrations of toxic byproducts.
Metabolism
Pyrroline 5 carboxylase synthase catalyses the first two steps in the biosynthesis of proline from glutamate. Proline is known to be accumulated in response to water deficit or salinity stress (Hanson & Hitz 1982) in plants but has also been observed in protozoa, eubacteria, algae (Delauney & Verma 1993) and marine invertebrates (Willett & Burton 2002). Acting as a cellular osmolyte, higher transcription of this gene in H families could reflect enhanced ability to maintain a more stable intracellular environment.
We observed higher levels of transcription of d-lactate dehydrogenase in L oysters, and this may reflect a shift towards increased anaerobic metabolism in L families. Although anaerobic metabolism is activated by marine bivalves during times of stress (Anestis et al. 2007), it is expected to yield less energy than aerobic metabolism. In France, oyster families that are susceptible to summer mortality are characterized by higher levels of reproductive investment than resistant oysters (Samain et al. 2007). If the consequence of being sensitive to stress is enhanced production of proteins that mediate cellular immunity and other processes, as is suggested by our data and those of Lang (2008), it is possible that metabolic exhaustion could contribute to mortality in L families. Although we did not study these genes with RT-QPR, the higher transcription of ribosomal proteins observed in the AFLP dataset is consistent with comparatively higher protein synthesis by L families.
Cell signalling
Protein tyrosine phosphatases (PTP) are involved in a variety of physiological processes such as signal transduction, cell cycle regulation and differentiation (Tonks & Neel 2001), through their role in regulating the reversible phosphorylation of tyrosine residues in proteins. This gene seems to be induced by bacterial infection, but only in L families. In this study, the inducible nature of the PTP genes transcription patterns contrasts with the constitutive transcription of the Rho GTPase gene. Members of the Rho subfamily are believed to be involved in the reorganization of the cytoskeleton but also in transcriptional regulation and cell division (Van Aelst & D’Souza-Schorey 1997). While both genes are involved in a plethora of processes, PTP gene transcription 12 h post-infection may reflect the rapid induction of metabolic regulation in L oysters. Because of the contradictory nature of RT-QPCR results vs. cDNA-AFLP for Rho GTPase, further investigation is needed to confirm the observed trend. However, it is interesting to note that previous studies have demonstrated that pathogenic microbes can subvert host cell integrity by targeting the host cell’s cytoskeleton (Gruenheid & Finlay 2003).
Conclusion
We report an investigation into the transcriptome response of the Pacific oyster (C. gigas) to infection with V. tubiashii using cDNA AFLP differential display. Our data indicate that oysters that are sensitive to heat stress may have a global enhanced sensitivity to bacterial infection (V. tubiashii) and tend to transcribe immune genes and ribosomal proteins at higher levels. These results confirm those of Lang (2008). In addition, oysters with high stress tolerance may have greater detoxification capacity. Hypothetically, L families have relatively more protein turnover and energy expended during maintenance metabolism, which could lead to less energy available for mounting stress responses (Hawkins et al.1989). Overall, our data support the hypothesis that stress resistance in Pacific oysters could be largely because of pre-stress differences in ‘general vigour’ that are maintained after infection rather than to differences in induced responses at the transcriptome level. Ultimately, a significant outcome of the study is the identification of transcripts and their functional roles, which enhances our understanding of the processes that contribute to differential survival in the context of summer mortality syndrome. Differential transcription seems to be indicative of genetically based variation in resistance to environmental stress. Genes linked to immune defence, and to a larger extent to detoxification capacity, provide both basic insights into oyster biology and candidate genes for further study, with the aim of enabling marker-aided selection to improve Pacific oyster survival in a context of summer mortality.
Acknowledgement
This research was supported with funding from the USDA Agricultural Research Service Shellfish Genetics Program (CRIS Project #5358-31000-001-00D).
Mandatory USDA-ARS Disclaimer Statement
Any use of trade, firm or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the United States Department of Agriculture or the Agricultural Research Service of any product or service to the exclusion of others that may be suitable.

