

Genetic model of MS severity predicts future accumulation of disability
Abstract
No genetic modifiers of multiple sclerosis (MS) severity have been independently validated, leading to a lack of insight into genetic determinants of the rate of disability progression. We investigated genetic modifiers of MS severity in prospectively acquired training (N = 205) and validation (N = 94) cohorts, using the following advances: (1) We focused on 113 genetic variants previously identified as related to MS severity; (2) We used a novel, sensitive outcome: MS Disease Severity Scale (MS-DSS); (3) Instead of validating individual alleles, we used a machine learning technique (random forest) that captures linear and complex nonlinear effects between alleles to derive a single Genetic Model of MS Severity (GeM-MSS). The GeM-MSS consists of 19 variants located in vicinity of 12 genes implicated in regulating cytotoxicity of immune cells, complement activation, neuronal functions, and fibrosis. GeM-MSS correlates with MS-DSS (r = 0.214; p = 0.043) in a validation cohort that was not used in the modeling steps. The recognized biology identifies novel therapeutic targets for inhibiting MS disability progression.
1 INTRODUCTION
The International Multiple Sclerosis (MS) Genetic Consortium (IMSGC) identified and validated over 200 MS susceptibility genes (Patsopoulos et al., 2017; Sawcer et al., 2011), but thus far has failed to validate genetic variants associated with MS severity (George et al., 2016; Sawcer et al., 2011). These negative results suggest that mechanisms that predispose subjects to develop MS largely differ from those that mediate central nervous system (CNS) destruction (i.e., disease severity) and that the latter remain undefined. The current study is based on the hypothesis that while there are genetic influences on the rate of CNS destruction (likely on the side of effector immune responses, susceptibility of CNS tissue to injury and its ability to repair), the effect sizes of common genetic variants identified in genome-wide association studies are too small to be reliably detectable using insensitive clinical outcomes. Additionally, distinct mechanisms may drive destruction of CNS tissue in different MS patients, as suggested by pathological heterogeneity of acute MS lesions (Lucchinetti et al., 2000). Thus, aggregating several candidate common genetic variants in models that allow complex, including nonlinear, interactions between alleles and accommodate heterogeneity in disease mechanisms has a greater potential for success.
MS severity can be defined as the speed of accumulation of neurological disability and is traditionally measured by two outcomes based on the broadly available Expanded Disability Status Scale (EDSS) (Kurtzke, 1983), but differ in the measurement of the time-aspect of MS severity. The older MS Severity Score (MSSS) (Roxburgh et al., 2005) uses MS disease duration as a measurement of time, whereas the newer age-related MS severity score (ARMSS) (Manouchehrinia et al., 2017) uses age. Unfortunately, multiple investigators have observed that MSSS and ARMSS do not predict future disability progression rates in moderately sized MS cohorts (Confavreux & Vukusic, 2006; Weideman et al. 2017a), likely because EDSS is a discrete scale ranging from 0 to 10, which cannot reliably measure individualized disability progression rates in intervals shorter than 10 years. Using machine learning, we developed an MS severity outcome called the MS Disease Severity Scale (MS-DSS) (Weideman et al., 2017a) based on the data-optimized, continuous Combinatorial Weight-Adjusted Disability Scale (CombiWISE) (Kosa et al., 2016), ranging from 0 to 100), which also adjusts disability progression slopes for therapeutic effects of applied treatments. By explaining a much larger proportion of variance of future disability progression rates, MS-DSS has more sensitivity in detecting biological modifiers of MS severity in comparison to MSSS or ARMSS. Consequently, we asked whether MS-DSS can be used to develop and validate a single-nucleotide polymorphism (SNP)-based Genetic Model of MS Severity (GeM-MSS) in a small, prospectively acquired and densely phenotyped longitudinal cohort of MS patients.
Because this cohort is underpowered for discovery research, we used a candidate gene approach with genes previously linked to MS severity, with poor reproducibility of individual variants in independent validation cohorts (Isobe et al., 2016; Jokubaitis & Butzkueven, 2016; Muhlau, Andlauer, & Hemmer, 2016; Sadovnick et al., 2017).
As a machine-learning algorithm of choice, we selected the random forest (RF), which captures complex interactions between alleles and disease heterogeneity, to model a total of 113 candidate genetic variants against MS-DSS in the training cohort. We validated the optimized model in a validation cohort that was not used in any aspect of modeling. While the validated GeM-MSS explained only a small proportion of MS severity variance, the described approach is applicable to larger cohorts that will allow for screening of significantly larger numbers of genetic variants.
2 SUBJECTS AND METHODS
2.1 Study population
In a blinded fashion, we genotyped 426 prospectively acquired subjects evaluated under natural history protocol “comprehensive multimodal analysis of neuroimmunological diseases of the CNS” (ClinicalTrials.gov Identifier NCT00794352). The study was approved by the Combined Neuroscience Institutional Review Board of the NIH, and all patients signed written informed consent forms. Upon unblinding diagnostic categories, 299 genotyped patients had a confirmed diagnosis of MS based on the 2010 revisions of McDonald's MS diagnostic criteria (Polman et al., 2011). Based on quality control (QC) filters described below, the final MS cohort was randomly split into training (N = 205) and validation (N = 94) subcohorts balanced for race, age, gender and family history of MS. The demographic and clinical data of these subcohorts was compared between diagnostic groups using an analysis of variance followed by Tukey's test with a Holm adjustment for multiple comparisons (Table 1).
| RR-MSa | SP-MSb | PP-MSc | |
|---|---|---|---|
| Training (n = 205) | |||
| n | 86 | 42 | 77 |
| Females/males | 54/32 | 21/21 | 39/38 |
| Age, years | 43.99 (18.01–76.43) | 54.28 (31.24–73.82)e | 57.94 (27.61–74.66)e |
| Disease duration, years | 13.91 (4–44) | 26.52 (9–49)e | 17.38 (6–45)d,f |
| MS-DSS | 1.44 (0.56–3.42) | 2.43 (0.87–5.19)e | 2.30 (0.34–4.90)e |
| MSSS | 2.83 (0.16–9.57) | 6.21 (1.43–9.82)e | 6.64 (0.64–9.85)e |
| Validation (n = 94) | |||
| n | 47 | 28 | 19 |
| Females/males | 30/17 | 10/18 | 11/8 |
| Age, years | 43.39 (24.95–65.44) | 53.44 (22.02–68.04)d | 59.19 (34.90–69.68)e |
| Disease duration, years | 14.51 (3–33) | 23.32 (8–33)d | 19.39 (8–46)e |
| MS-DSS | 1.61 (0.51–2.31) | 2.49 (0.81–5.33)d | 2.22 (0.81–4.43)d |
| MSSS | 2.98 (0.26–8.75) | 6.98 (1.7–9.56)e | 6.71 (2.55–9.56)e |
- Note. Continuous data are shown as the mean of all measurements and the minimum and maximum values are in parentheses.
- aRelapsing-remitting MS.
- bSecondary progressive MS.
- cPrimary progressive MS.
- dp < 0.05 when compared to RR-MS using one-way ANOVA with Holm adjustment for multiple comparisons.
- ep < 0.0001 when compared to RR-MS using one-way ANOVA with Holm adjustment for multiple comparisons.
- fp < 0.0001 when compared to SP-MS using one-way ANOVA with Holm adjustment for multiple comparisons.
2.2 Genotyping and quality control
SNP genotyping was performed using the Illumina HumanOmniExpress v.1.0 Neuro-X array on EBV-transformed peripheral blood mononuclear cells (PBMCs) and whole-blood extracted DNA samples using the standard protocol recommended by the manufacturer (Illumina, San Diego, CA). The OmniExpress Neuro-X array is an Illumina Infinium iSelect HD Custom Genotyping array containing >710,000 markers and an additional 24,706 custom variants designed for neurological disease studies. Of the custom variants, approximately 12,000 are designed to study Parkinson's disease and are applicable to both large population studies of risk factors and to investigations of familial diseases and known mutations (Nalls et al., 2015).
We used the Genotyping Analysis Module within Genome Studio v.1.9.4 to perform sample and variant QC checks. The threshold call rate for sample inclusion was 95%. QC of sample handling was determined by comparing the reported sex with genotypic sex estimated from X chromosome heterogeneity. X chromosome heterogeneity calculations were based on common SNPs from the International HapMap Project (The International HapMap Consortium et al., 2007). Samples considered heterozygosity outliers (>±3 standard deviations [SDs] from the sample mean) or with discrepancies between reported sex and genotypic estimated sex were excluded. All 299 samples used in the analysis passed QC.
Additional variant QC checks were performed in PLINK v.1.9. We excluded variants with a minor allele frequency (MAF) ≤5%, missingness ≥5%, and genotypes that deviated from Hardy–Weinberg equilibrium (p < 1 × 10−5). Of the 720,107 genotyped autosomal variants, 595,556 variants passed all genotyping QC filters.
2.3 Genotype imputation
Data was prepared for imputation using HRC-1000G-check-bim.pl v.4 (http://www.well.ox.ac.uk/~wrayner/tools/#Checking) and PLINK v.2 Imputation was done on the Michigan imputation server, using the Haplotype Reference Consortium (HRC) reference panel version r1.1,2016 (http://www.haplotype-reference-consortium.org) on unphased data. Importantly, given that our cohort includes samples of African-American ancestry, this panel has been shown to estimate genotypes for common variants in African-American participants with high imputation quality (Vergara et al., 2018). Molecular genotyping of HLA-DRB1 alleles was provided by the NIH blood bank as a clinically approved test and reported to investigators via medical records. We removed variants with an imputation quality score <0.5 or an MAF <0.05 as additional QC measures.
2.4 Strategy for identification of candidate SNPs
We used the following publicly available databases to identify literature that reported variants associated with MS severity: Ensembl (Zerbino et al., 2018), Phenotype–Genotype Integrator (Ramos et al., 2013), GWAS Catalog (MacArthur et al., 2017), and PubMed. Studies were selected based on the criteria that supplementary material was publicly available and that the objective of the study was discovery rather than replication. We used “multiple sclerosis severity,” “multiple sclerosis age of onset,” and “genome-wide association studies” to identify literature sources that reported genetic associations with MS severity. Previously published studies used three general strategies to measure MS severity: (1) MSSS, (2) age of disease onset, or (3) destructiveness of CNS tissue measured by magnetic resonance imaging (MRI). We included studies that used any of these three strategies. We excluded copy number variants, insertions, and deletions, and we required that the reported association p-value be <10−5 for this analysis.
2.5 Modeling outcome MS-DSS and additional assessment outcomes: MSSS and ARMSS
MS-DSS (Weideman et al., 2017a) is assigned by a statistical model using gradient boosting machines. MS-DSS includes disability measured by a highly sensitive CombiWISE (Kosa et al., 2016), mathematically adjusted for the efficacy of administered treatments using a published formula (Weideman, Tapia-Maltos, Johnson, Greenwood, & Bielekova, 2017b), the amount of CNS-tissue destruction measured by the Combinatorial MRI Scale of CNS Tissue Destruction (COMRIS-CTD) (Kosa et al., 2015), and additional features of lower variable importance, including demographic data. The model uses the following cross-sectional data, listed in order of statistical importance: (1) therapy-adjusted CombiWISE divided by patient age (CombiWISE/age); (2) CombiWISE; (3) COMRIS-CTD; (4) time to first therapy, which measures the delay (in years) from disease onset to initiation of treatment; (5) difference in therapy-adjusted and measured CombiWISE, which reflects the variant of the disease that is treatable by current immunomodulatory treatments; (6) age, and (7) family history of MS. MS-DSS, the modeling outcome in the current study, is automatically calculated from user-inputted raw data via a web interface (https://bielekovalab.shinyapps.io/msdss).
Even though we previously determined that MSSS (Roxburgh et al., 2005) and its later modification, ARMSS (Manouchehrinia et al., 2017), were too insensitive to predict future rates of accumulation of disability in moderately sized MS cohorts (Weideman et al., 2017a), we assessed the correlation between GeM-MSS and these widely used MS severity scales as sensitivity analyses.
2.6 Random forest (RF)-based Genetic Model of MS Severity (GeM-MSS)
The RF algorithm (Breiman, 2001; Hastie, Tibshirani, & Friedman, 2009) is a highly successful ensemble learning method suited for high-dimensional data (such as genomics) that aggregates many individual decision trees. A decision tree is a modeling approach used in classification and regression problems that utilizes several features (e.g., laboratory tests) to classify an outcome (e.g., presence or absence of a disease or level of disease severity) by finding the optimal split (e.g., a concentration of an analyte) for each “branch” of the decision tree. The main problem of tree-based classifiers is an “overfit” of the data, making predictions from the classifiers unstable. RF partially mitigates this problem by averaging together results from multiple decision trees (often thousands) that are constructed using bootstrapped samples of the training data, with observations not used to build the tree forming an “out-of-bag” (OOB) group. RF further alleviates the “overfit” problem by introducing an element of random selection in features considered when performing splits (i.e., the algorithm only considers a random subset of features for every split in the tree-building process). The main tuning parameters in an RF are the number of trees to grow (ntree) and the number of variables to sample for each node split (mtry). In the current study, trees were grown until the OOB error stabilized and the default mtry was used (approximately 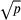 , where p is the total number of available features). Briefly, the OOB error is a measure of the RF model accuracy that is derived from the OOB samples, and the final OOB error estimate for the model is the mean prediction error on each sample using only the predictions from trees where the sample was in the OOB group. Because the RF models still have the potential to overfit the training data, we used the iterative process adapted from previous studies (Calle, Urrea, Boulesteix, & Malats, 2011; Gregorutti, Michel, & Sainte-Pierre, 2017) where the least important variables ranked by permutation variable importance (Breiman, 2001) were removed and the RF was rebuilt until the OOB error increased by more than 1% (Figure 1). To ensure the stability of the variable importance results, 30 individual RF models using different random seeds were constructed and were averaged together at each iteration. The final set of predictive variants were identified as the variants remaining in the model before the OOB error increased. The predictions from the OOB error-stabilized model, referred to henceforth as GeM-MSS, were assessed in a validation cohort that was not used in the model building. In all analyses, we used the implementation of the RF algorithm from the ranger package (Wright & Ziegler, 2017) in R v.3.4.4 (Core Team, 2018) that has been adapted for high-dimensional datasets.
, where p is the total number of available features). Briefly, the OOB error is a measure of the RF model accuracy that is derived from the OOB samples, and the final OOB error estimate for the model is the mean prediction error on each sample using only the predictions from trees where the sample was in the OOB group. Because the RF models still have the potential to overfit the training data, we used the iterative process adapted from previous studies (Calle, Urrea, Boulesteix, & Malats, 2011; Gregorutti, Michel, & Sainte-Pierre, 2017) where the least important variables ranked by permutation variable importance (Breiman, 2001) were removed and the RF was rebuilt until the OOB error increased by more than 1% (Figure 1). To ensure the stability of the variable importance results, 30 individual RF models using different random seeds were constructed and were averaged together at each iteration. The final set of predictive variants were identified as the variants remaining in the model before the OOB error increased. The predictions from the OOB error-stabilized model, referred to henceforth as GeM-MSS, were assessed in a validation cohort that was not used in the model building. In all analyses, we used the implementation of the RF algorithm from the ranger package (Wright & Ziegler, 2017) in R v.3.4.4 (Core Team, 2018) that has been adapted for high-dimensional datasets.

2.7 Data and code availability
All custom code developed and used in this manuscript are available in supplementary file 1. Genetic and phenotype data are available in the database of Genotypes and Phenotypes (dbGaP, https://www.ncbi.nlm.nih.gov/gap).
3 RESULTS
3.1 Identification of candidate SNPs
The information about the genotyped SNPs and their published sources are summarized in Supplemental Table 1. In total, 116 SNPs were identified for association with MS severity. After QC, 113 variants located in the vicinity of 95 unique genes remained for analysis.
3.2 Development of RF genetic model (GeM-MSS) and its optimization based on variable-importance data
We applied the RF technique to model MS-DSS based on 113 SNPs previously reported for association with MS severity (Figure 1). The model was built and optimized in the training cohort, while its general validity was tested in the validation cohort balanced for race, age, gender, and family history of MS (Table 1). Because of the high likelihood that some (perhaps the majority) of the tested 113 SNPs represent “noise,” we adopted an iterative process of discarding the least important SNP in each subsequent iteration of the RF model and evaluated the performance of this simplified iteration using the OOB error of the RF. This iterative process of model optimization stopped after observing at least a 1% increase in the error of predicting the OOB samples. This indicates that all remaining genetic variants are important in predicting MS severity of all subjects in the training cohort. The resulting GeM-MSS had 19 remaining variants. Compared to the model with all 113 variants, the OOB error of the GeM-MSS decreased by 14.4% (initial OOB error = 1.059 vs. GeM-MSS OOB error = 0.907), even though the root mean squared error (RMSE) in both models was similar (initial RMSE = 0.444 vs. GeM-MSS RMSE = 0.464). We observed a strong and statistically significant correlation between the model predicted and measured MS-DSS in the training cohort (Figure 2a; r = 0.969; p = 8.76 × 10−125).

Because machine learning techniques have the potential to overfit the training data, we tested the validity of the model in a cohort that was not used in any step of the modeling. In this validation cohort, we observed a statistically significant correlation, although of much lower strength, between the model predicted and observed MS-DSS (Figure 2b; r = 0.214; p = 0.043).
3.3 Assessment of GeM-MSS with MSSS and ARMSS
Because MS-DSS is a complex model that uses data that may not be available for all MS cohorts, we evaluated the ability of GeM-MSS to predict MS severity outcomes that were not used for its development but are available for genotyped MS cohorts that belong to other investigators.
Thus, we assessed the correlation between GeM-MSS and MSSS and ARMSS in the training and validation cohorts. We observed a moderate correlation when comparing GeM-MSS to MSSS (Figure 3a; r = 0.578; p = 2.71 × 10−19) and ARMSS (Figure 3b; r = 0.579; p = 4.20 × 10−19) in the training data. In the smaller validation cohort, we observed positive, although nonsignificant associations between GeM-MSS and EDSS-based MS severity scores (Figure 3c; r = 0.202; p = 0.056 for MSSS and Figure 3d; r = 0.165; p = 0.120 for ARMSS).

3.4 Biological interpretation of validated GeM-MSS
To obtain biological interpretation of GeM-MSS, the variable importance ranking, MAF, and nearest gene of the 19 remaining variants are given in Table 2. In total, the GeM-MSS outcome predicted by the 19 variants explains approximately 4.4% of the variance in MS-DSS. Each of the variants remaining in GeM-MSS is relatively common and had an MAF greater than 0.10. These variants are within the vicinity of 12 genes, including YWHAG, XYLT1, CAMK2D, and KDM2B. Three genes (YWHAG, XYLT1, PVRL2) were represented by several SNPs and represent regions of high linkage disequilibrium (LD) within the genes. The most important variant in the model (rs11765693) is located in the YWHAG gene and is succeeded by two variants in the XYLT1 gene (rs12927173 and rs2059283) (Figure 2c). Interestingly, the signals in the XYLT1 and YWHAG genes are among the most significant associations to MS severity in each of the discovery cohorts that initially identified these associations (Supplemental information, References 23 and 24, respectively). This result provided evidence that efforts to replicate the top genetic associations to MS severity may be improved, in part, by using MS-DSS as an outcome.
| Chromosome | Nearest gene | Variant | Type | Alleles | MAF | Model rank |
|---|---|---|---|---|---|---|
| 1 | SLAMF7 | rs35967351 | Intronic | A/T | 0.25 | 16 |
| 4 | CAMK2D | rs987694 | Intronic | G/A | 0.34 | 5 |
| 6 | ARID1B | rs7744583 | Intronic | G/A | 0.33 | 10 |
| 7 | C1GALT1 | rs10259085 | Intronic | C/T | 0.50 | 11 |
| MAGI2 | rs246462 | Intronic | A/G | 0.34 | 8 | |
| YWHAG | rs11765693 | Intronic | A/G | 0.29 | 1 | |
| rs17149161 | Intronic | C/A | 0.29 | 4 | ||
| rs7779014 | Intronic | C/T | 0.29 | 6 | ||
| rs7789940 | Intergenic | A/G | 0.29 | 9 | ||
| 8 | CSMD1 | rs9644362 | Intronic | G/C | 0.18 | 13 |
| PSD3 | rs7015570 | Intronic | A/G | 0.21 | 14 | |
| 12 | KDM2B | rs7134248 | Intronic | T/C | 0.48 | 7 |
| 16 | CDH13 | rs4315313 | Intronic | T/C | 0.43 | 12 |
| XYLT1 | rs2059283 | Intronic | T/G | 0.48 | 3 | |
| rs12927173 | Intronic | T/C | 0.48 | 2 | ||
| 19 | PVRL2 | rs4803766 | Intronic | G/A | 0.48 | 17 |
| rs2972566 | Intronic | G/C | 0.48 | 15 | ||
| rs419010 | Intronic | C/T | 0.48 | 18 | ||
| rs394221 | Intronic | C/T | 0.48 | 19 |
Several selected variants are predominantly expressed in the CNS (especially in neurons), such as tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein gamma (YWHAG), membrane-associated guanylate kinase, WW and PDZ domain containing 2 (MAGI2), CUB, and Sushi multiple domains 1 (CSMD1). CSMD1 and MAGI2 variants have been associated with cognitive dysfunction; whereas, YWHAG, which belongs to the 14-3-3 family of proteins, participates in glutamate-induced cell death. These neuronal variants had high variable importance in the GeM-MSS model. In addition to its neuronal role, CSMD1 also inhibits formation of the complement membrane attack complex in the CNS and, therefore, may prevent CNS tissue destruction (Kraus et al., 2006).
Two variants in the vicinity of xylosyltransferase I (XYLT1) had the second and third highest variable importance. XYLT1 catalyzes the first step in biosynthesis of glycosaminoglycans, including chondroitin sulfate proteoglycans, which have been shown to impede remyelination (Lau et al., 2012). Finally, several variants with high variable importance (i.e., MAGI2, YWHAG, XYLT1) participate in or regulate the process of epithelial–mesenchymal transdifferentiation, which is linked to fibrosis. Therefore, restructuring the extracellular matrix into the gliotic scar may be a crucial limiting step for CNS repair in MS.
On the side of the immune system, the selected variants highlight the importance of cytotoxic cells (T cells and natural killer [NK] cells) and their interaction with B cells and plasma cells. The cross-linking of the signaling lymphocytic activation molecule family 7 (SLAMF7) enhances interferon-γ production and cytotoxicity (Comte et al., 2017) of NK cells and T cells. SLAMF7 is also expressed on plasma blasts, plasma cells, and activated B cells (Llinas et al., 2011), possibly mediating costimulatory functions of these cells of humoral immunity for CD8+ T cells and NK cells via homotypic interactions. Finally, SLAMF7 is also highly expressed on macrophages, where it mediates phagocytosis (Chen et al., 2017) and thus may contribute to myelin stripping in MS. On the other hand, poliovirus receptor-related 2 ([PVRL2], also called herpesvirus entry mediator B and NECTIN2 or CD112) binds to a pair of negative (i.e., T-cell immunoglobulin and ITIM domain [TIGIT]) and positive (i.e., CD226 [DNAM-1]) regulators of NK and cytotoxic T-cell functions (Stein, Tsukerman, & Mandelboim, 2017). These immune-related variants had lower variable importance in comparison to the CNS-enriched variants.
4 DISCUSSION
Genetic modifiers of the MS disease course remain elusive. In fact, it is hard to even estimate how much of the MS severity variance is genetically determined. Intuitively, a reasonable estimate of the genome-wide genetic contribution to MS severity might be up to 50%, considering the effect of treatments, environment, and stochastic processes. The previous attempts to link MS susceptibility variants to MS severity were not successful (Isobe et al., 2016; Jokubaitis & Butzkueven, 2016; Muhlau et al., 2016; Sadovnick et al., 2017). If these results were true negatives, then genetic variants that predispose patients to acquiring MS under favorable environmental conditions did not influence the rate of CNS tissue destruction or its recovery. This conclusion is counterintuitive because therapeutic success of Food and Drug Administration–approved immunomodulatory treatments on accumulation of MS disability (Weideman et al., 2017b) leaves no doubt that the immune system partakes in CNS tissue destruction. At least some of the MS susceptibility variants linked to dysregulated immune responses would be expected to also influence destruction of CNS tissue. Therefore, we considered it likely that previous negative results were, to a certain extent, because of Type II errors. This conclusion is strongly supported by GeM-MSS, which contains SNPs linked to effector immune mechanisms known to be associated with destruction of varied human tissues, including CNS, such as complement-mediated cell lysis (i.e., formation of the terminal membrane attack complex) and cellular cytotoxicity.
Validation of GeM-MSS also support our hypothesis that common genetic variants are unlikely to exert a strong negative influence on CNS tissue destruction or its recovery. Rather, complex and often nonlinear relationships between immune-related MS susceptibility genes and genes expressed predominantly in CNS tissue likely mediate susceptibility versus resistance of CNS tissue to injury or affect recovery mechanisms such as remyelination or synaptogenesis. The variable importance metrics provide strong support to CNS-driven mechanisms such as glutamate-induced cell death and neurodegenerative processes previously linked with cognitive dysfunction, which may affect neurogenesis and CNS repair in general. Another pathogenic process that GeM-MSS identified is the restructuring of extracellular matrix in a form of fibrotic scar that may prevent remyelination and possibly also new synaptogenesis. Since in other organs fibrosis is often a consequence of chronic inflammation, we consider it likely that this process is immune related in MS as well.
Finally, we would like to discuss technical aspects of our and other published studies; aggregating effects of multiple variants into a single genetic model with greater predictive power is intuitive and at least partially supported by published literature (Pan et al., 2016). Modeling 116 validated MS susceptibility variants in a cohort of 125 early MS cases followed for 5 years, Pan et al. (2016) developed an additive model (Cumulative Genetic Risk Score [CGRS]) consisting of seven MS susceptibility SNPs. If the patient had more than two of the seven risk genotype variants, CGRS showed a dose–response relationship with MS severity measured by annualized change in EDSS; their model explained 32.7% of variance in disability progression. However, the publication of CGRS did not include validation in an independent cohort. GeM-MSS selected only one the seven variants from CGRS (rs35967351; SLAMF7). When we attempted to validate the CGRS dose–response relationship in our combined training and validation cohorts that is almost 3 times larger than the training cohort used for modeling CGRS, we observed no evidence of a linear relationship (Supplemental information). Instead, we saw a slight, nonsignificant increase in the measured MS severity in subjects with at least three risk alleles when compared to two or less risk alleles using several MS severity outcomes, including the sensitive MS-DSS.
It is unrealistic for seven frequent genetic risk alleles to explain 32.7% of the variance in disability progression. This would imply unusually high effect sizes, which should have been easily identified/validated in previous and much larger studies (George et al., 2016; Sawcer et al., 2011). Thus, the performance of CGRS, derived from a small, unvalidated cohort, represents an overfit (Ioannidis, 2008). The strong model performance in the training cohort should not be perceived as an automatic guarantee of the model's clinical utility. Instead, an independent validation cohort, that did not contribute in any way to feature selection or model development, is an absolute requirement for assessing the true value of any model. The correlation coefficient of 0.969 in the training cohort indicates that even the final GeM-MSS model grossly overestimates the true relationship between the 19 risk alleles and MS severity in the training cohort. The performance of GeM-MSS in the validation cohort, demonstrating mild statistical significance and explaining less than 10% of MS severity variance, is much more credible. Yet, the obtained p-value of 0.04 indicates a 4% chance of validating these findings in a similar cohort if no relationship is present. Therefore, we would welcome an independent validation of GeM-MSS from investigators with larger genomic/clinical datasets. We consider the probability that GeM-MSS can be validated by independent investigators that own large cohorts of genotyped MS patients with linked EDSS to be high. We base this belief on the fact that GeM-MSS correlates significantly with EDSS-based MS severity scores that did not contribute to model development in the training cohort, and that even in the small validation cohort we observed a positive association between GeM-MSS and MSSS and ARMSS.
In conclusion, this study provides a genetic model of MS severity that aggregates several previously identified common genetic variants and provides important genetic insight into MS disability progression. The introduced technical advances (MS-DSS, combining SNPs into a single model that captures nonlinear effects and disease heterogeneity) can be used to further improve GeM-MSS through multicenter collaborations. Only such a collaborative assembly of densely genotyped and phenotyped data can perform genome-wide search of additional variants contributing to MS severity.
ACKNOWLEDGMENTS
The study was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) and the Clinical Center/U.S. National Institutes of Health (NIH). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
K.C.J. performed the analysis, quality control of the genetic data, and generated the figures; K.S. performed the initial genetic data quality control checks; C.B. conceived and designed the scripts for the automated RF pipeline; K.C.J. and B.B. drafted the manuscript; P.K and M.T. prepared the samples for genotyping; P.K maintained the database of clinical and demographic data; D.H. performed the genotyping and imputation; A.M.W wrote the script and computed the therapy-adjusted values required for MS-DSS. B.B. conceived the study and oversaw the analysis. All authors reviewed the manuscript.
ETHICAL APPROVAL
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Open Research
DATA AVAILABILITY STATEMENT
All custom code developed and used in this manuscript are available in the supplementary information files. Genetic and phenotype data are available in the database of Genotypes and Phenotypes (dbGaP).

