

Habitat differentiation vs. isolation-by-distance: the genetic population structure of Elymus athericus in European salt marshes
Abstract
We investigated genetic differentiation among populations of the clonal grass Elymus athericus, a common salt-marsh species occurring along the Wadden Sea coast of Europe. While E. athericus traditionally occurs in the high salt marsh, it recently also invaded lower parts of the marsh. In one of the first analyses of the genetic population structure in salt-marsh species, we were interested in population differentiation through isolation-by-distance, and among strongly divergent habitats (low and high marsh) in this wind- and water-dispersed species. High and low marsh habitats were sampled at six sites throughout the Wadden Sea. Based on reciprocal transplantation experiments conducted earlier revealing lower survival of foreign genotypes we predicted reduced gene flow among habitats. Accordingly, an analysis with polymorphic cross-species microsatellite primers revealed significant genetic differentiation between high and low marsh habitats already on a very small scale (< 100 m), while isolation-by-distance was present only on larger scales (60–443 km). In an analysis of molecular variance we found that 14% of the genetic variance could be explained by the differentiation between habitats, as compared to only 8.9% to geographical (isolation-by-distance) effects among six sites 2.5–443 km distant from each other. This suggests that markedly different selection regimes between these habitats, in particular intraspecific competition and herbivory, result in habitat adaptation and restricted gene flow over distances as small as 80 m. Hence, the genetic population structure of plant species can only be understood when considering geographical and selection-mediated restrictions to gene flow simultaneously.
Introduction
Genetic population differentiation between sites and habitats has been the rule rather than the exception in many genetic and demographic studies on plant populations (see Hamrick & Godt 1991 for a review). Geographic differentiation results from limited dispersal in many plant species and is conveniently summarized in isolation-by-distance models, first proposed and formalized by Wright (1946). Which specific life history traits of a given species on one hand, and which attributes of the environment on the other impede or promote dispersal (and gene flow) is relatively well understood (e.g. Slatkin 1985, 1987; Hansson 1991; Bohonak 1999; Cain et al. 2000). In plants, short ‘realized dispersal’ (i.e. the successful establishment of a plant at the site), in conjunction with the limited physical distances that propagules (spores or seeds) travel, often does not allow gene flow over larger distances (Cain et al. 2000). Consequently, increasing geographical distance will lead to increasing genetic isolation of populations.
In terrestrial plants, the main obstacles for long-distance dispersal are geographical barriers such as mountain ranges, lakes, rivers, or shorelines. Conversely, in aquatic plants, the latter three of these barriers are the dispersal pathways for hydrochorous dispersal. Not much is known about the quantity of hydrochorous dispersal and realized dispersal distances (but see Koutstaal et al. 1987). Salt-marsh plants are interesting for studying dispersal because they are intermediates between terrestrial and aquatic plants. They are terrestrial in terms of their morphology, physiology and ecology, while most species are dispersed by water (although additionally also by wind or animals, Bakker et al. 1985). In our study species, Elymus athericus (Link), Gould (sensuVan der Meijden 1990), the shed seeds will float in the water for various amounts of time and may be transported over long distances by tidal currents. Ten per cent of the propagules of our study species still float after 30 days of immersion in sea water (J. P. Bakker, unpublished data). We hypothesized that a common and rapidly colonizing species such as E. athericus should have effective dispersal and would therefore show little genetic population differentiation through isolation-by-distance. Elymus athericus is one of the most common plant species in the Wadden Sea salt marshes, sometimes covering more than 70% of the area (e.g. on Schiermonnikoog, the Netherlands, 53°30′, 6°10′). Since E. athericus has increased considerably in the past three decades, leading to a concomitant decline in species diversity in many marshes (Leendertse et al. 1997; Van Wijnen et al. 1997), we were also interested to infer colonization speed and abilities from marker data.
Genetic habitat differentiation has been neglected as a potential barrier to gene flow in many population genetic studies. Adaptation of populations to locally varying conditions has mostly been approached by transplantation or common environment experiments as reviewed by Endler (1986) and Van Andel (1998). Habitat adaptation becomes apparent through significant genotype by environment interaction or home site advantage. In contrast, DNA-based molecular markers have been primarily used for studying neutral population genetic processes such as drift and gene flow, although recently, there has been increasing interest in marker-based approaches of habitat differentiation in plants (Nevo et al. 1994; Li et al. 2000b; Engelen et al. 2001). The two habitats studied here are the low and the high salt marsh, which differ in numerous biotic and abiotic factors, such as shore height, inundation and vegetation composition (Adam 1993). In a parallel study we found that biotic factors, particularly the grazing pressure of herbivores (geese, hares) were more important for genetic habitat differentiation in E. athericus than abiotic factors, such as inundation frequency (Bockelmann et al. 2002). Reciprocal transplantation experiments with different life-history stages, both seedlings and adult ramets demonstrated the heritability of habitat-specific phenotypic traits. These differences in the phenotype are indicative of fitness differences among plants originating from high and low marsh in this species (Bockelmann 2002). We therefore predict that fitness differences in plants from different origin may reduce the gene flow from one population to another (Stanton et al. 1997; Vekemans & Lefebvre 1997). Such restrictions to gene flow will be reflected by genetic differentiation at molecular marker loci such as microsatellites, although the markers themselves are selectively neutral. We hypothesized that the described (Table 2) differences in the life history between the low and the high marsh populations have repeatedly resulted in different selection regimes restricting gene flow. As a consequence we expected to find genetic differentiation in neutral DNA markers at replicated sites across the Wadden Sea area.
| Source of variation | d.f. | Density (shoots m−1)* | Shoot length† | Spikelets per spike† | Ramet number† |
|---|---|---|---|---|---|
| Habitat | 2 | Low < High | Low < High | Low > High | Low > High |
| Site | 5 | SNK, HS < KD, 4th, WD, WHV | KD, 4th, WD, WHV < SNK, HS | KD, 4th, WD, WHV < SNK, 4th, WD, WHV < HS | WHV > SNK, 4th > KD, WD, HS |
| Habitat × Site | 2 | not significant | not significant | not significant | not significant |
| Residual d.f. of anova | 108 | 331 | 331 | 331 | |
| n | 10 | 30 | 30 | 30 |
- * April 1998.
- † August 1998.
- KD = Kobbeduin, 4th = 4th-creek, WD = Willemsduin, SNK = Sönke-Nissen-Koog Vorland, WHV = Westerhever, HS = Helmsand, Low = Low marsh, High = High marsh.
The genetic population structure of most European salt marsh plants, including E. athericus, is still unknown (but see Baumel et al. 2001). In this study, we directly investigated the realized exchange of propagules between populations of E. athericus with cross-species microsatellite markers. To assess the significance of habitat-correlated genetic differentiation on a broader scale, we replicated the comparison of low and high marsh habitats at several locations as advised by Endler (1986) and Prentice et al. (1995). This is the first study where the consistency of genetic habitat differences was tested over six sites on a landscape scale (see Prentice et al. 2000 for an experimental approach of small-scale habitat differentiation).
Materials and methods
Elymus athericus
Elymus athericus[Sea Couch, synonymous taxonomic names are, e.g. Elymus pycnanthus (Godron) Melderis, Elytrigia pungens (Pers.) Tutin, or Agropyron pycnanthum (Godron) Godron & Gren] is a tall, clonal grass native to salt marshes. It occurs along the North Sea coast between southern Denmark and the southern Netherlands but can also occur further south. It is wind pollinated, as are most Poaceae. The main dispersal units are spikelets, which bear up to five seeds (A.-C. Bockelmann, personal observation). Spikelets have no obvious dispersal mechanism. Breeding experiments demonstrated that this species is hexaploid (6x = 2n = 42; Stace 1995; Hess et al. 1998). Elymus athericus is usually outcrossing (Dewey 1983), but is also self-compatible (Bockelmann 2002). Selfing does not lead to reduction in seed viability (Bockelmann 2002). Depending on site and habitat, the population morphology and phenotype of E. athericus can be very different. Higher on the marsh, we mainly find continuous meadows, whereas populations are patchy lower on the marsh (Table 1). At our study sites, there were pronounced differences in the plant phenotypic traits. Mean shoot density in the high marsh is higher and shoots are longer. The production of spikelets per spike and new ramets is higher in the low marsh (Table 2). Differences in plant phenotypes were consistent across all six sites, i.e. there was no significant statistical interaction among ‘site’ and ‘habitat’.
| Site | Habitat | Population morphology | Shore height* (cm + MHW) | Inundation frequency year−1† | Age of marsh (years)‡ | Grazing by livestock§ | Resident herbivores* | N-mineralization (kg ha−1 yr−1)*¶ |
|---|---|---|---|---|---|---|---|---|
| Kobbeduin | high | meadow | 40–98 | 125 | 125 | until 1973 | none | no data |
| low | meadow | 22–38 | 270 | 6–? | none | hare, geese | no data | |
| 4th-creek | high | meadow | 83–128 | 20 | 86 | until 1958 | hare | no data |
| low | patches | 50–76 | 115 | 6–? | none | hare, geese | no data | |
| Willemsduin | high | meadow | 60–102 | 40 | 25 | none | hare, rabbit | 3.5¶ |
| low | patches | 57–86 | 90 | 13 | none | hare, geese | no data | |
| Helmsand | high | meadow | 65–81 | no data | no data | until 1986 | 26.0** | |
| low | patches | none | geese | no data | ||||
| Westerhever | high | meadow | 50–120 | 120 | no data | until 1984 | 55.0†† | |
| low | meadow | 10–20 | 170 | none | geese | 32.0†† | ||
| Sönke-Nissen- | high | meadow | 34–55 | 96 | 56 | until 1988 | 30.8‡‡ | |
| Koog-Vorland | low | patches | 38 | 5–10 | none | geese | 66.6‡‡ |
- Geese are Branta bernicla bernicla and B. leucopsis, the hare is Lepus europaeus and the rabbit is Orynctolagus cuniculus.
- * A.-C. Bockelmann, unpublished data.
- † Bockelmann et al. (2002).
- ‡ Olff et al. (1993).
- § J.P. Bakker, unpublished data.
- ¶ Van Wijnen et al. (1999).
- ** R. Neuhaus, unpublished data.
- †† Bockelmann & Neuhaus (1999).
- ‡‡ Dahl (2001).
Study sites and sampling design
Developing a sampling design to study genetic population structure and gene flow in Elymus athericus was difficult because we had no information on clone size and dispersal distance prior to the present analysis. To ensure that we included the relevant spatial scales, at which genetic differentiation is partitioned in this species, we sampled E. athericus populations on three different scales. To test for differentiation by geographical distance we chose six sites along the Wadden Sea coast in Germany and on the Dutch Island of Schiermonnikoog, which were between 2.5 and 443 km distant from one another (Fig. 1). Additional differences between sites were the age of the marsh, the land-use history and the nitrogen mineralization rate (as a measure of nitrogen availability and productivity, Table 1). At each site we recognized two subpopulations, one higher on the shore, where E. athericus forms continuous almost monospecific meadows (hereafter named ‘high’). The second subpopulation was situated lower on the marsh, where E. athericus forms patches within vegetation dominated by Puccinellia maritima and Festuca rubra (hereafter named ‘low’). The locations at different shore heights on the elevational gradient are associated with pronounced differences in inundation frequency and other ecological factors (Table 1). In each subpopulation we sampled leaf material of three individual ramets in 10 blocks to include two levels of potential clone size. The sampling distance between tillers was 1.5–3 m; the distance between blocks was 10–30 m. The total sampling size was 360. The total sampling area (approximately 5000 m2 per population) was chosen according to the geomorphology or the management regime at the site to ensure potentially unrestricted gene flow. The sampled leaf material was preserved through drying in tubes with silica gel until DNA extraction.

The study area, indicating three sites in Germany and three on the Dutch Island of Schiermonnikoog.
Cross-species modification of microsatellites, sequencing, and segregation patterns
We used microsatellite primer pairs originally developed for two other species of Poaceae, Elymus caninus (Sun et al. 1998a) and Triticum aestivum (Röder et al. 1998). Out of 26 primer pairs tested (including 12 primer pairs of E. alaskanus; Sun et al. 1998b), five amplified and showed consistently scorable results for E. athericus (Table 3). These were used in the present study.
| Primer name | Repeats in original species | Repeats in Elymus athericus‡ | Allele size range (bp) | No. of alleles | Fluorescent label |
|---|---|---|---|---|---|
| WMS2* | (CA)18 | (CA)10§ | 180–295 | 9 | 6-FAM (blue) |
| WMS6* | (GA)40 | (GA)15 | 140–164 | 11 | NED (yellow) |
| WMS44* | (GA)28 | no data | 115–144 | 4 | 6-FAM (blue) |
| ECGA22† | (CT)27 | (CT)10 | 128–156 | 4 | HEX (green) |
| ECGA89† | (GA)17 | (GA)12G(GA)4 | 197–207 | 4 | HEX (green) |
- * Röder et al. (1998).
- † Sun et al. (1998a).
- ‡ Complete sequences can be found in GenBank Accession No. AJ517215-25.
- § Not present in all individuals.
Alleles of four of the five primers derived from other species were subsequently characterized by cloning and sequencing according to standard protocols (one primer had to be left out for logistic reasons and time constraints also prevented the development of species-specific primers). Alleles in our study are operationally defined as amplified fragments originating from one of the five primer pairs.
Sequences contained microsatellites in both E. caninus- and Triticum aestivum-derived primers (Table 3). For the latter primers in particular, changes in the nonrepeat region suggested the multiple presence of a microsatellite locus, with subsequent loss of the microsatellite at some of the loci. The occurrence of additional, nonmicrosatellite fragments has also been reported by Stachel et al. (2000). Our data also indicate that null-alleles are present. Six polymerase chain reaction (PCR) products (i.e. corresponding to a hexaploid genome) were only found occasionally in primer WMS6, whereas the average number of alleles is two for WMS44, WMS2 and ECGA89 and three for WMS6 and ECGA22, prohibiting a co-dominant analysis of data. The size range of the amplified fragments in E. athericus differed from the species for which the primers were originally designed (Table 3).
We investigated qualitatively whether the amplified microsatellite fragments would segregate in offspring, using semi-artificial pollination. For this purpose, two groups of ramets from the field (distance 20 m) were dug out and two inflorescences, one of each ramet group, were placed together in a paper bag (mesh width < 0.2 mm, n = 4). We analysed the parent genotypes and five seeds of each spike. All fragments present in the offspring were also present in the putative parents (Fig. 2). In all cases, amplified fragments present in either parent were absent in at least one offspring. We could thus infer that the primers used are inherited in a Mendelian way.

Segregation pattern of microsatellite peaks of four primers (ECGA22, green 115–156 bp, ECGA 89, green, 197–207 bp, WMS6, black, 115–144 bp, WMS44, blue, 138–164 bp) in Elymus athericus. The two upper panels show the parent plants, the two lower panels shows two seeds from the mother. The x-axis gives the size of the peak in base pairs and the y-axis indicates the relative concentration of the amplified peak or microsatellite fragment.
Microsatellite genotyping
For microsatellite genotyping, genomic DNA was extracted from 5–10 mg of dry leaf tissue using the Qiagen plant extraction kit (Qiagen). One microlitre (containing ∼5–10 ng) of E. athericus DNA-extract was subjected to PCR with fluorescently labelled forward primers (and unlabelled backward primers). A 10-µl reaction contained 1-µl Promega Buffer B (10×, Promega Corp.), 200 µm of each dNTP, 1.5 µm MgCl2, 5 µm of each primer, 0.1% (wt/vol) bovine serum albumin, and 0.25 units Promega Taq polymerase. Amplification conditions were: 3 min hot start at 94 °C, followed by 10 cycles of 1 min denaturation at 94 °C, 1 min annealing at 50–60 °C, and 30 s extension at 72 °C. The following 20–23 cycles had the same conditions except a 5 °C lower annealing temperature and an extension time of 20 min for the last cycle.
For visualization on an ABI-377 automated sequencer (Applied Biosystems), the PCR products of primer pairs WMS6, WMS44 and ECGA22, and of primer pairs WMS2 and ECGA89, respectively, were pooled and size scored against an internal lane standard. Positive and negative controls were always included to check the repeatability of the results.
Data analysis and statistics
Amplified fragments were analysed using the genescan and genotyper software packages (Applied Biosystems). It follows from the DNA sequence data (see above) that the calculation of single-locus based allele frequencies in E. athericus is not possible. Consequently, we chose to analyse fragment frequencies as multilocus fingerprints in which each allele was either scored present or absent. We only analysed alleles with a frequency > 10% in at least one population.
Genotypic diversity
The genotypic or clonal diversity of a population was calculated as PD, the proportion of detectable genotypes (Ellstrand & Roose 1997), which gives the fraction of number of genets (= clones, g) detected and ramets (r) sampled (PD = g r−1). Differences in genotypic diversity between populations from different sites were tested by contingency table test among all six populations.
Analysis of molecular variance
Between-population differentiation was partitioned into habitat-correlated and geographical effects using analysis of molecular variance (amova; Weir & Cockerham 1984; Excoffier et al. 1992; for fingerprint data). An amova was also used to calculate pairwise population differences as ΦST. For this purpose, we have to assume that inbreeding coefficients are constant over populations, an assumption that is reasonable given the wind-pollination and likely predominance of outcrossing in E. athericus. Prior to all analyses on population differentiation, duplicate multilocus genotypes were excluded from each population (42 samples in total). We used the computer program tfpga (Miller 1997) for nested and unnested amova, and confirmed the results independently in an unnested amova in genetix (version 3.3, Belhir et al. 1996, 98). Because of the dominant/recessive inheritance of the data obtained by partially degenerate microsatellites in a hexaploid species, heterozygosities could not be calculated.
Genetic diversity
Genetic diversity within and among populations was calculated using Shannon's diversity measure (Lewontin 1972). This measure has the advantage that it does not assume Hardy–Weinberg equilibrium as amova does. This assumption may not be met in the salt-marsh environment because of nonrandom mating, e.g. because of the patchy population structure, or within clone inbreeding. This makes it a useful tool for comparative purposes in the analysis of genetic population structure next to amova. The Shannon index was calculated according to Pielou (1975) as:
H′ = −Σ pi ln * pi
where pi is the frequency of the ith microsatellite peak (= locus). We chose the natural logarithm to calculate H′, but any log base may be adopted (Magurran 1983; p. 35). We followed Chalmers et al. (1992) to detect within-population differences of H′ depending on the primer used by averaging across primers:

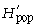 was calculated for each locus separately and subsequently averaged over all loci of each primer. The overall diversity of all populations (the ‘species’, sp.)
was calculated for each locus separately and subsequently averaged over all loci of each primer. The overall diversity of all populations (the ‘species’, sp.)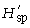 can be calculated as
can be calculated as 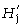

with p̄s being the average frequency of peaks of one primer in all samples (318 plants in our study). It follows that  is the component of diversity within stands of one population and that
is the component of diversity within stands of one population and that 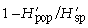 is the component of diversity between populations (Chalmers et al. 1992).
is the component of diversity between populations (Chalmers et al. 1992).
Isolation-by-distance
We explored how much differentiation between population pairs can be explained by geographical distance. Because in E. athericus, long-distance dispersal of seeds is most likely by water (Bockelmann et al. 2002), the geographical distance between the populations was measured along the coastline (excluding bays such as the Dollard).
Apart from an overall correlation, we also considered high and low populations separately to test the hypotheses that founder effects would increase the variance in differentiation among low populations. In each data set, the relation of pairwise genetic distances (assessed as ΦST) with geographical distance was tested with a Mantel test (1967) using genetix.
Results
Genotypic and genetic diversity
Populations in different habitats did not differ in the number of distinguishable genotypes (d.f. = 5, χ2 = 0.529, P = 0.99; Table 4). Across all populations the number of identical genotypes ranged from 0 to 17%. If there were any similar multilocus genotypes they occurred within blocks (within the three plants sampled 1–3 m distant). The genetic diversity in Elymus athericus populations (expressed by the Shannon index H′) ranged from 1.41 to 4.07 (Table 4). Twenty-nine per cent of the total genetic diversity was partitioned between populations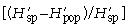 . Paired t-tests revealed no differences in genetic diversity between primers, habitats, or sites.
. Paired t-tests revealed no differences in genetic diversity between primers, habitats, or sites.
| Site | Habitat | Genotypic diversity PD | Genetic diversity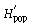 |
|---|---|---|---|
| Kobbeduin | High | 1.00 | 2.79 |
| Low | 0.93 | 2.56 | |
| 4th-creek | High | 0.83 | 2.28 |
| Low | 0.75 | 1.79 | |
| Willemsduin | High | 0.88 | 2.54 |
| Low | 0.96 | 3.60 | |
| Helmsand | High | 0.97 | 3.23 |
| Low | 1.00 | 1.41 | |
| Westerhever | High | 0.96 | 3.51 |
| Low | 0.90 | 2.65 | |
| Sönke-Nissen- | High | 0.96 | 4.07 |
| Koog-Vorland | Low | 0.89 | 3.36 |
| Average |
2.82 | ||
 |
4.10 | ||
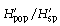 |
within | 0.72 | |
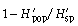 |
between | 0.29 |
- Genotypic diversity = proportion of distinguishable genotypes, PD, Genetic diversity = Shannon index for the genetic diversity
-
within population
 = genetic diversity calculated for
= genetic diversity calculated for
-
each population separately,= genetic diversity calculate
-
across all samples,= within population genetic
-
diversity,= between population genetic diversity.
Population differentiation between sites and habitats
We found significant genetic differentiation of amplified fragment frequencies between sites and habitats. A one-way amova revealed that 14% of variance was distributed among all 12 subpopulations. When partitioning geographical and habitat effects using hierarchical amova we found that 8.9% (d.f. = 5; 95% CI: 4.3–14.6%) of the variance could be explained by site and 14% (d.f. = 6; 95% CI: 8.4–21.9%) by the two chosen habitats (residual d.f. = 274). The differentiation between habitats was thus 64% higher than between sites despite the small geographical distance (maximally 80 m). The amount of genetic variance explained together by site (8.9%) and habitat (14%) in amova is approximately the same amount as found with the Shannon index (29%, see above).
Of all the amplified fragments 94% occurred in at least 75% of the high marsh populations. In the low marsh 88% of the fragments were present in at least 75% of the populations. None of the fragments occurred in less than 50% of the populations.
Pairwise population differentiation and isolation-by-distance
We calculated pairwise population differentiation in ΦST between the 66 pairs of populations in our study (Table 5, Fig. 3). The values ranged between 0.009 and 0.315, with a significant differentiation (P < 0.05) for 59 population pairs. Between habitats within one location, all but two comparisons (KD Low vs. KD High, WHV Low vs. WHV High, Table 5) showed significant fixation indices. These populations had a patchy morphology whereas the populations without differentiation between low and high marsh were continuous populations (Table 1).
| KD low | 4th high | 4th low | WD high | WD low | HS high | HS low | SNK high | SNK low | WHV high | WHV low | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| KD high | 0.040 | 0.073 | 0.111 | 0.061 | 0.175 | 0.310 | 0.118 | 0.190 | 0.096 | 0.213 | 0.153 |
| KD low | 0.077 | 0.092 | 0.074 | 0.177 | 0.321 | 0.127 | 0.218 | 0.154 | 0.211 | 0.172 | |
| 4th high | 0.106 | 0.009 | 0.057 | 0.144 | 0.057 | 0.175 | 0.119 | 0.172 | 0.126 | ||
| 4th low | 0.132 | 0.190 | 0.315 | 0.141 | 0.280 | 0.185 | 0.267 | 0.214 | |||
| WD high | 0.073 | 0.192 | 0.069 | 0.158 | 0.112 | 0.167 | 0.142 | ||||
| WD low | 0.081 | 0.106 | 0.197 | 0.176 | 0.220 | 0.203 | |||||
| HS high | 0.171 | 0.295 | 0.294 | 0.307 | 0.266 | ||||||
| HS low | 0.146 | 0.044 | 0.122 | 0.082 | |||||||
| SNK high | 0.117 | 0.021 | 0.016 | ||||||||
| SNK low | 0.145 | 0.089 | |||||||||
| WHV high | 0.016 |
- KD = Kobbeduin, 4th = 4th-creek, WD = Willemsduin, HS = Helmsand, SNK = Sönke-Nissen-Koog Vorland, WHV = Westerhever, Low = Low marsh, High = High marsh.

Genetic differentiation (expressed by the FST estimator ΦST) between population pairs of Elymus athericus at different geographical distances. FST values at ∼80 m distance indicate differentiation between habitats (populations at low or high shore height in the marsh within single sites, grey circles), values at larger distances (∼100 m to 500 km) show site differentiation (filled circles indicate high marsh, open circles indicate low marsh population pairs).
We found weak but significant isolation-by-distance through pairwise comparison of population pairs among all sampled locations. Pairwise ΦST values were significantly correlated with geographical distances. However, the coefficient of determination was low. (R2 = 0.095, Pmantel = 0.04, 5000 permutations). When testing the isolation-by-distance among populations belonging to each habitat separately correlation was almost significant for all six high populations (Pmantel = 0.063, 5000 permutations, R2 = 0.60) but clearly nonsignificant for all low ones (Pmantel = 0.4, 5000 permutations, R2 = 0.004). At between-sites distances < 10 km, pairs of low marsh sites showed a higher differentiation than expected under the isolation-by-distance model (Fig. 3). At larger distances (between 60 and 443 km) genetic differentiation increased with distance. The average differentiation among habitats within sites of 14% (amova, see above) corresponds to a differentiation solely through geographical distance of approximately 150 km.
Surprisingly, the Helmsand population was more similar to the populations on Schiermonnikoog, although it is situated in a clayey mainland marsh in the northeastern Wadden Sea such as Sönke-Nissen-Koog-Vorland and Westerhever (Table 1, Fig. 3).
Discussion
The use of cross-species microsatellite primers compared to other molecular markers
The cross-species microsatellite primers successfully used in this study in Elymus athericus were adapted from Triticum aestivum and Elymus caninus. Trials with other plant species have had mixed results (e.g. Van Treuren et al. 1997; White & Powell 1997; Streiff et al. 1998; Reusch 2000; Van der Velde 2001). Our results are consistent with studies where primers of E. caninus have successfully been used for other Elymus species (Sun et al. 1998a; 1998b). The success might be based on the close relationship of species within the tribe Triticeae, as suggested in the light of difficult morphological discrimination and unsolved taxonomic classification (Dewey 1983).
Cross-species microsatellites might be an alternative to the costly and time-consuming development of species-specific microsatellites. Even if insertions and deletions of parts of the repeat region prohibit co-dominant analysis as in our case, species specificity, better repeatability (Gillet 1999) and interpretation (less complex allele pattern) make it superior to random priming techniques such as random amplified polymorphic DNA or amplified fragment length polymorphism. In E. athericus the application of traditional allozyme techniques is difficult. Using this technique, we analysed the same 12 populations as in this study and found that the results are flawed by a bad repeatability and temporal variation of allozyme pattern within genotypes (Bockelmann 2002). The data derived from allozyme analysis might have overestimated genetic variation, in particular between high and low marsh. The explanation may be physiological differences in enzyme activity on a small spatial and temporal scale and even within individuals (clones) as discussed earlier by Clark & Koehn (1991). Furthermore, co-dominant interpretation is also prohibited for allozyme data by the complex banding pattern of this hexaploid species. We propose that allozyme data should, if used at all, be interpreted phenotypically rather than genetically in this species. In other Elymus species allozymes are applicable but show lower levels of polymorphism than microsatellites (Sun et al. 1999; Sun et al. 2001).
Genetic differentiation and diversity in comparison with other Elymus species
The among-population differentiation ranged between 0.089 and 0.14 (FST estimator ΦST) in E. athericus. Much stronger genetic differentiation between populations has been reported from allozyme or random amplified polymorphic DNA analysis of other Elymus species (GST values ranging from 0.55 to 0.95). However, in contrast to the study species E. athericus, these taxa were at least predominantly, if not exclusively, self-pollinating [Elymus alaskanus, Diaz et al. 1999; Elymus fibrosus, Diaz et al. 2000; Elymus glaucus,Knapp & Rice (1996), all 4x, 2n = (28)]. Stronger population differentiation has been predicted for self-pollinating as opposed to outcrossing species (Hamrick & Godt 1991).
Geographic isolation and dispersal
In contrast to our expectations of strong genetic exchange by means of water currents, differentiation and partitioning of genetic diversity between populations was already significant on a scale of < 100 m. Geographic distances of more than 60 km led to weak but significant isolation-by-distance in Elymus athericus, with considerable variance introduced by habitat-correlated genetic differences (see below). If we assume that E. athericus seeds or spikelets can float for 30 days on the water (J. P. Bakker, unpublished results) and further assume a maximal water velocity of 3.7 km/h of the Wadden Sea tidal current at spring tide (Dietrich et al. 1975), the potential dispersal distance might be approximately 600 km within 1 week. This is much more than the average distances from one marsh to another. However, these dispersal distances are probably overestimates. Spikelets must first of all leave the creek system and reach the Wadden Sea because velocities within creeks are much lower (0.72 km/h, Winskowsky 1998) than in open water and not always orientated towards the tidal flats, as could be shown in two pilot studies on Schiermonnikoog (A.-C. Bockelmann and T. Wels, unpublished results). Moreover, tidal currents change direction with every tidal cycle, and wind direction and wind speed may be more important in determining dispersal direction (Koutstaal et al. 1987). Nevertheless, potentially substantial gene flow between the geographical sites seems to be possible.
Rather than limits to propagule dispersal, demographic processes such as low germination rates of E. athericus can probably explain the difference between potential and realized dispersal distances. When addressing the differentiation between sites in a study on Schiermonnikoog, Bockelmann (2002) could show that germination is probably the most critical life history stage in E. athericus. Rare successful long-distance dispersal events could lead to strong founder effects in new populations. This could explain that the differentiation between sites is stronger than expected based on the potential dispersal rates. That founding events promote genetic divergence in E. athericus is also apparent from the surprisingly high genetic differentiation among young, newly founded E. athericus populations in low areas that is much stronger than would be expected through isolation-by-distance.
Habitat differentiation as barrier to gene flow
Although distances among high and low marsh habitats are short, we observed repeatedly even more differentiation between habitats within sites than between sites up to 443 km away from each other. As such, our results confirm for the first time that genetic habitat differentiation over short geographical distances (maximally 80 m) can be consistent over several sites on a landscape scale. The hierarchical sampling allowed the distinction of habitat differentiation on one hand and isolation-by-distance on the other. The results imply consistent differences in biotic or abiotic factors between the low and high habitat at all six studied sites that result in different local selection regimes for E. athericus.
Three mechanisms could explain the differentiation of microsatellite patterns between habitats through selection. First, there could be direct selection on microsatellite loci. This is very unlikely because in eukaryotes, microsatellites are thought to be situated predominantly in noncoding introns and contain no transcribed genetic information (Hancock 1999; but see Li et al. 2000a, 2000b for a discussion of possible direct selection on microsatellite loci driving habitat divergence in wild emmer wheat Triticum dicoccoides). Moreover, direct selection would lead to consistent frequency changes of specific PCR fragments of the loci under selection. While this has been demonstrated for habitat-correlated differences at allozyme loci (Prentice et al. 1995; Schmidt et al. 2000), it is probably not the case here. In E. athericus 91% of the alleles are present in at least 75% of the populations, while no PCR fragment occurs in less than 50% of the populations. The commonness of fragments can serve as an argument against the second possible mechanism, genetic linkage of microsatellite loci with functional genes.
Instead, we propose that genetic differentiation of plant traits, as a result of selection on the life-history traits of E. athericus, resulted in limited gene flow. Ecological habitat adaptations of life cycle stages, such as the establishment of seedlings, have been shown experimentally to limit realized gene flow (Bockelmann 2002). In parallel transplant experiments, we have shown that resistance to herbivores, and competitive ability towards neighbouring conspecific plants differs among genotypes from high and low habitats. We found that seedlings with parents from the high marsh were less adapted to stronger herbivory present in the low marsh, while seedlings with low marsh parents were less able to survive competition in the high marsh. This corresponds with the intensity of herbivory, respectively, the strength of competition in these habitats (Van de Koppel et al. 1996; Bockelmann 2002). Consequently, the establishment of seedlings from nonlocal populations without herbivory or competition in E. athericus is limited (Bockelmann 2002). The history of domestic livestock grazing, inundation frequency and nitrogen mineralization (Table 1) differ markedly not only between habitats but also between sites and may further contribute to genetic divergence in the phenology of E. athericus populations. It may result in differences in the timing of reproduction, the period of flowering, seed shedding or the timing of germination.
In summary, we have shown that neutral molecular markers repeatedly revealed restricted gene flow between low and high salt-marsh populations despite high potential propagule transport by water and wind. Hence, the genetic population structure of plant species can only be understood when considering geographical and selection-mediated restrictions to gene flow simultaneously.
Acknowledgements
We want to thank two anonymous reviewers for comments on the manuscript and Silke Carstensen, Catherine Schmuck and Roos Veeneklaas for their technical assistance. A.-C.B. is grateful for the support of the Max-Planck-Society through W. Lampert. A.-C.B. was funded by the Hans-Böckler-Stiftung and an Ubbo-Emmius grant of Groningen University.
References
Anna-Christina Bockelmann was a PhD student at the Centre for Ecological and Evolutionary Studies (Biology Centre, University of Groningen, the Netherlands) supervised by J. P. Bakker and R. Bijlsma. The molecular work in this study was carried out in co-operation with T. B. H. Reusch from the Max-Planck-Institute for Limnology in Plön, Germany. The aim of A.-C.B.'s PhD thesis was the analysis of the invasion of Elymus athericus in European salt marshes from an ecological and evolutionary perspective.

