

The Many Chemists Who Could Have Proposed the Woodward-Hoffmann Rules But Didn't: The Organic Chemists Who Knew of the Smoking Guns**
This paper is Paper 7 in the 27-paper series on the history of the development of the Woodward-Hoffmann rules, also known at the Principle of the Conservation of Orbital Symmetry. For Paper 6, see: J. I. Seeman, Chem. Rec. 2022, 22,1–67; doi.org/10.1002/tcr.202200065.
Abstract
It is a reasonable question to ask, why, as of 1965 when the five Woodward-Hoffmann communications appeared, did no other organic chemist discover the orbital symmetry rules for pericyclic reactions? The previous two papers in this 27-paper series on the history of the Woodward-Hoffmann rules discussed the physical chemists, chemical physicists, and theoretical chemists who could have solved the pericyclic no-mechanism problem; and the organic chemists in whose laboratory many of the key hints to this problem were found but still did not solve the problem. The stories of 16 other chemists who knew of (at least portions of) the pericyclic no-mechanism problem are presented in this paper. Social, political, and scientific explanations are presented as partial rationalizations as to why none of these individuals – except Woodward with Hoffmann – solved the pericyclic no-mechanism problem.
“I was given the honor of introducing Woodward and Hoffmann at an ACS meeting (1973) where they were receiving the first Cope Award, largely for orbital symmetry. I remember remarking ruefully to someone at the meeting that those of us who did not win the Cope Award could console ourselves with the thought that it took two guys to beat us.”1 – Jerome A. Berson (2010)
“… the unfortunate rift that exists today between physical and organic chemistry. Few chemists have a real knowledge of both subjects – and incidentally fewer still have also a knowledge of quantum theory. Yet a knowledge of all three is essential for a proper understanding of organic chemical theory.”2 – Michael J. S. Dewar (1949)
1. Introduction
“Being at the right place at the right time” is axiomatic to success in science
John D. (Jack) Roberts, the noted physical organic chemist, member of the US National Academy of Sciences, recipient of the US National Medal of Science (the highest civilian award in the United States) and the Priestley Award (the highest award of the American Chemical Society (ACS)), and many other awards, entitled his autobiography, The Right Place at the Right Time. He was quite correct in his choice of title, for Jack was often at the right place at the right time. But ironically, despite all his successes and awards, he didn't take advantage of every opportunity. And, of course, who does?
There were two opportunities that Jack Roberts missed, in which he was in exactly at the right place at the right time, each of which led to Nobel Prizes for other chemists. One was the 2003 Nobel Prize in Physiology or Medicine that was awarded to Pau C. Lauterbur and Peter Mansfield “for their discoveries concerning ‘magnetic resonance imaging.’” Roberts was one of the first organic chemists to apply nuclear magnetic resonance spectroscopy (NMR) for the study of structure and reaction mechanisms. Lauterbur applied NMR to develop magnetic resonance imaging (MRI).
Roberts's other missed opportunity was determining the mechanism of pericyclic reactions. As will be discussed below, Roberts had most of the prerequisites to solve that problem. He was an expert in small ring chemistry. He was an expert in molecular orbital theory and computation. He had great graduate students during this time who were experts in physical organic chemistry, e.g., George Whitesides and Joseph B. Lambert. He had access to the necessary laboratories and state-of-the-art computers. But he did not solve the problem.
Nor did any of the many other chemists who were in the right place at the right time.
Why was it not any of the many other organic chemists, all well-educated and experienced chemists who had mastered much of the breadth of organic chemistry? This question was answered in the previous article in this series for the organic chemists in whose laboratories some of the mysterious reactions – reactions for which there was no mechanism available – were first reported. In this paper, an assembly of organic chemists will be discussed. While none of chemists experienced firsthand the discovery of any of these mysterious reactions, they were almost certainly knowledgeable of some of these reactions and had other experiences that placed them in an otherwise perfect place to have discovered orbital symmetry control. In a sense, case studies for many individual chemists will be presented. Each case study provides insights into the workings of one chemist. These case studies as a group paint a picture of the academic organic chemistry community in the early 1960s.
This and the two previous papers in this series3, 4 have two overarching themes. One, of course, is the presentation of a series of mini-stories, one for each chemist who was right there and could have but did not solve the pericyclic no-mechanism problem. The second theme shared by the papers in this series is a biographical picture of chemistry in the mid-to-late 1950s and 1960s.
Note: Throughout this paper, the terms “electrocyclization,”5 “sigmatropic rearrangement,”6 and “pericyclic reaction”7 will be used often. However, these terms were not invented until Woodward and Hoffmann did so in 1965 for the first two and 1969 for the last term. As this paper discusses chemistry pre-1965 chemistry, terms such as valence isomerization and cycloaddition will be used herein, just as they were used in the pre-1965 literature. It is worth noting, as Arthur C. Cope did in 1952, that in 1934 Baker suggested the term “valence tautomerization” for reactions better known today as electrocyclizations.8
2. The First Set of Chemical Hints
An understanding of this paper requires knowledge of what the no-mechanism problem was as of January 1965. To stand in the shoes of chemists of that era would help in understanding the opportunities and complexities facing those chemists. A full description of the chemical events leading up to the 1965 Woodward-Hoffmann papers is presented in Paper 2 of this series.9 Papers 53 and 64 of this series also present the chemical background to the no-mechanism problem.
In the absence of further explanations of the no-mechanism in this paper, Figure 1 may be sufficient. Imagine yourself in 1964. Imagine that you are aware of the chemistry summarized in Figure 1. Imagine also that most of your peer group has not assembled the chemistry clues shown in Figure 1 together in one place, and perhaps you have not done so either, thus not fully appreciating the breadth and magnitude of the pericyclic no-mechanism problem. That is what faced the organic chemistry community in the early1960s.

Specific instances of pericyclic no-mechanism reactions as of January 1965. (eq. 1 and eq. 2) Pairs of stereospecific reactions, thermal versus photochemical. Provitamin D and lumisterol photolyze to previtamin D while isopyrocalciferal and pyrocalciferol photocyclize to photoisopyrocalciferol and isopyrocalciferol, respectively. On the other hand, previtamin D photocyclizes to provitamin D and lumisterol; but previtamin D thermally cyclizes to isopyrocalciferol and pyrocalciferol. (eq. 3) [2+2] will photocyclize to a cyclobutane but will not do this conversion thermally unless there are several highly polar substituents. The cyclization then is nonconcerted. (eq. 4) But the [4+2] cycloaddition will occur thermally (the Diels-Alder reaction) while the photochemical [4+2] is rare.10-14 (eq. 5) Thermal cyclobutene ring openings results in the less stable (E,Z)-1,4-disubstituted-1,3-butadienes.15, 16 (eq. 6) The Cope rearrangement (see Section 3.3) is an example of a sigmatropic rearrangement.
Away we go!
3. Organic Chemists Who Might Have Solved the Pericyclic No-Mechanism Problem
3.1. Preface
As mentioned above, what follows are case studies for 16 chemists with a specific focus on the aspects of their chemical background that might have helped them solve the no-mechanism problem. The case studies are as idiosyncratic as the chemists themselves.
3.2. Walter Karl Friedrich Bernhard Hückel (1895–1973)
Walter Hückel (Figure 2) was the elder brother of the physicist-turned-theoretical chemist Erich Hückel (1896–1980) who discovered in the early 1930s Hückel molecular orbital theory and published the distinction of 4n+2 and 4n electron systems as they pertain to stability. Walter was an eminent professor of organic chemist, having received several honorary doctorates and many awards (e. g., the Otto Wallach Plaque (Germany), the Lavoisier Medaille (France), the Grignard Medal (France) and the Medal of the Royal Akademy (Amsterdam)).17 According to SciFinder, he had 227 publications (Figure 3).

Walter Hückel.

Publication record of Walter Hückel (SciFinder).
Walter Hückel was a student of the Nobel laureate Adolf Windaus. His first publication in 1920 was on Baeyer's strain theory.18 His first independent publications were on thermodynamics of simple organic compounds.19-24 His interests then moved rather permanently into experimental research involving stereochemistry and reaction mechanisms. A very early success was his preparation and isolation of the two configurational isomers of decalin in 1923, considered by some the beginning of modern stereochemistry.17 His review in 1928 on Steric Hindrance revealed his interest in the effect of substituents on conformationally mobile systems,25 predating the conformational analyses of Derek Barton of the early 1950s.26, 27 In the late 1950s, together with his student Michael Hanack, he published several papers on Constellation Analysis,28, 29 an early synonym for conformational analysis. He was an expert in the classical reactions (e. g., SN2 and the Walden inversion30) and steric hindrance on ester hydrolyses31, 32 with a specific focus on monoterpenes (e. g., menthol and its stereoisomers, 21 papers on this topic from 192933 to 1960).34 In addition to his research accomplishments, Hückel published extensively on the history of chemistry and wrote many (short) obituaries. Perhaps Hückel's major accomplishment was his massive two-volume series on Theoretical Principles of Organic Chemistry which appeared in numerous ever-growing editions (several of which appeared in English as well as in German), the last being published in 1955.35
Hückel wrote in the 1935 edition that
“It is certain that an understanding of important chemical questions can only be gained on the basis of modern quantum theory, and it will therefore no longer be possible for organic chemists to ignore the latest developments in atomic and molecular physics… ”36
Nonetheless, even in the 1955 edition of his treatise,35 Hückel incorporated almost nothing of his brother Erich's MO theory. Rather, the textbook spoke mostly in the language of valence bond theory and resonance.37, 38 Had the two Hückel brothers communicated about the pericyclic no-mechanism problem – even about Vogel's 195439 or Criegee's 1959 cyclobutene ring openings (eq. 5),16 or had seen Havinga's 1961 paper,40 the history of chemistry may well have been considerably different. There is little evidence that there was much collaboration between the two brothers, though they did publish one paper together on The Theory of Induced Polarities in Benzene.41
3.2.1. Why and Why Not Walter Hückel
This is an example of possibilities forsaken. Of two brothers, one an experimental organic chemist, the other a theoretical physicist who made major contributions to the discipline of chemistry, together they could have easily solved the pericyclic no-mechanism problem. These two scientists essentially knew chemistry quite well, but perhaps not the latest advances (1950s and early 1960s state-of-the-art): Walter, as judged by the scholarship of his textbooks and the breadth of his publications; Erich, because he invented Hückel molecular orbital theory.
But by the early 1960s, neither was looking to solve a major problem in chemistry. Neither was looking for a breakthrough, over-the-top disciplinary challenge to solve by themselves let alone together. As mentioned in Paper 5 in this series,3 Erich Hückel had long retired from active scientific research. By the late 1950s, Walter Hückel was nearing retirement and finishing projects long underway. Furthermore, his publications were quite distant from the valence isomerizations and vitamin D thermal and photochemistry that we key to the solution of the pericyclic no-mechanism problem. Lastly, brother Erich was not interested in quantum chemical solutions to organic chemical problems, and certainly not molecular orbital expositions.
In sum, they both were from an earlier generation.
3.3. Arthur C. Cope (1909–1966)42
3.3.1. General Background Information
In this era of numerous octogenarian and nonagenarian chemists,43, 44 sadly two contemporaries of Woodward's died in their 50s, both of whom are discussed in this paper. One is Saul Winstein, discussed in Paper 6 of this series,4 and the other is Arthur C. Cope, who died in 1966 at the age of 55. Woodward died in 1979 at the age of 62, two years before Hoffmann received the Nobel Prize for the Woodward-Hoffmann rules. But Woodward did receive his Nobel Prize in 1965 “for his outstanding achievements in the art of organic synthesis.” Moreover, Woodward did receive one very major award for the Woodward-Hoffmann rules, that being the first Arthur C. Cope Award, jointly with Hoffmann in 1973 (Figure 4 and Figure 5).

(left to right) Hoffmann, Herman Bloch (Chair of the Board of Directors of the American Chemical Society), Mrs. A. C. Cope, and Woodward, at the first Arthur C. Cope Award presentation, 1973. Photograph courtesy Harvard University Archives.

Cover page of Jerome A. Berson's Arthur C. Cope Award dinner program. Berson introduced the two recipients of the first Cope Award, R. B. Woodward and Roald Hoffmann. Both award recipients autographed this program for Berson (upper right-hand corner, Hoffmann; middle, Woodward). Berson had been a Woodward student. And Berson and Hoffmann had become friends beginning with their correspondence in the mid-1960s.45 At that point, Berson had also published the most dramatic, supporting experimental data on orbital symmetry control of pericyclic reactions.46
Arthur Cope was a highly successful organic chemist.42 He received his Ph.D. in 1932 from the University of Wisconsin, studying with Samuel M. McElvain. He began his independent career with a National Research Council Fellowship with Emmett P. Kohler at Harvard during which he published several sole-author papers. Since most of Cope's career was at MIT, it is natural to associate the Cope reaction with MIT, but that is not the case. In fact, he first taught at Bryn Mawr (1934–1940 (Figure 6); with the summer of 1935 at Illinois) and four years at Columbia (1941–1945) before his years at MIT (1945–1966, at his death). He was chair of the Department of Chemistry at MIT for almost 20 years, which he ran with an iron hand. In fact, it was while he was at Bryn Mawr that the first papers on the Cope reaction were published.

Arthur C. Cope performing a classroom demonstration at Bryn Mawr College, ca. 1940. Photograph courtesy Bryn Mawr College and Allison Mills, College Archivist.
The 1939 Bryn Mawr College yearbook has an interesting comment about Cope. It reads,
“Mr. Cope, whose office increased in inaccessibility proportionally to the decrease in our chemical ability, now breezes cheerfully to the main floor of the New Science Building, where he performs mighty researches both day and night.”47
Cope led a very active professional life with a major focus on the ACS. He was President of the ACS in 1961, on the ACS Board of Directors for 16 years (1951–1966) and was Chair of the Board of Directors for seven of those years. Cope left half of his considerable estate, built by patent royalties, to the ACS from which the Cope Awards and the Cope Scholar Awards are generously funded. Indeed, the corpus of the Arthur C. Cope Fund grew so much that in 1986–13 years after the first Cope Award – the ACS instituted the Cope Scholar Award. The Cope Award consists of $25,000, plus a medallion and a certificate, and up to $2500 for travel expenses to the ACS Fall Meeting. The Cope Scholar Awards, of which ten are given annually, each consist of a $5000 prize, a certificate, and a $40,000 unrestricted research grant plus up to $2500 in travel expenses to the fall ACS National Meeting.
3.3.2. Gateway: Pericyclic Reactions
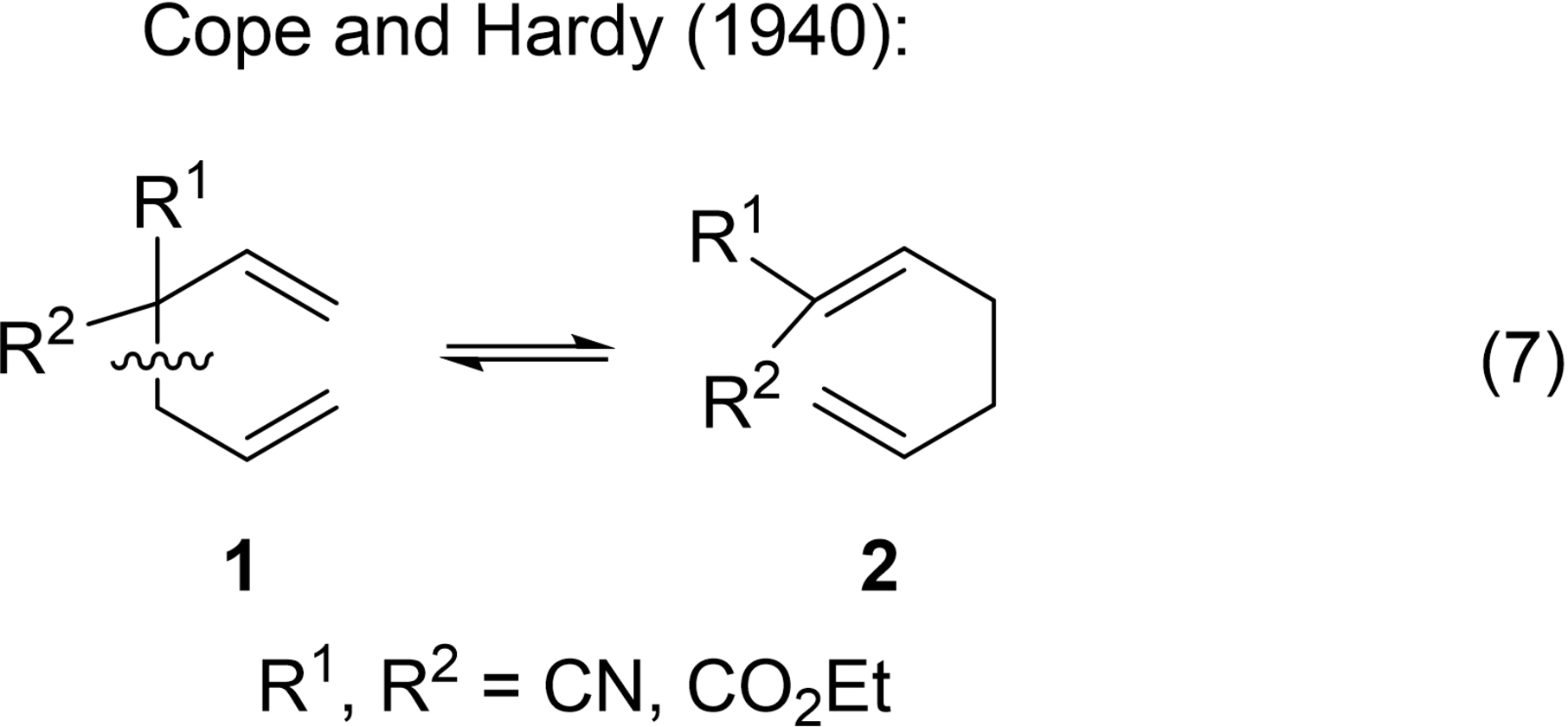
In 1940, Cope and Elizabeth M. Hardy (Figure 7) published a monumental serendipitous discovery, the Cope rearrangement (Figure 8),48 a prototypical sigmatropic rearrangement analyzed as such in the third Woodward-Hoffmann publication.6 In that era, principal investigators often identified their publications by the title of their research theme. Cope considered this paper with Hardy as Paper V in his series The Introduction of Substituted Vinyl Groups.48 Cope and Hardy pointed out that “it seems probable that this new rearrangement is similar in type to the Claisen rearrangement… and that it is motivated by similar forces.48 Figure 8, excerpted from Cope's first paper on this rearrangement, illustrates the electron flow using Robinson-type reaction arrows49 proposed by Cope in 1940.

In 1940, Elizabeth M. Hardy published the first paper on the Cope rearrangement with her Ph.D. advisor Arthur C. Cope at Bryn Mawr College.48 This photograph appears in the 1938 edition of the McGill University yearbook. Photograph courtesy McGill University Library and Greg Houston and Jessica Lang.

Graphical excerpt from Cope and Hardy's 1940 paper on the first Cope rearrangement48 characterized by Woodward and Hoffmann in 1965 as a sigmatropic rearrangement.[6] Reprinted (adapted) with permission from A. C. Cope, E. M. Hardy, J. Am. Chem. Soc. 1940, 62, 441-444. Copyright 1940 American Chemical Society.
In that paper, Cope and Hardy observed the transformation 1 → 2 and wrote,
“The most probable way in which [1] could rearrange into [2] [eq. 7] involves a shift of the allyl group from the alpha to the gamma position, accompanied by a shift of the double bond…48”
Later in their paper, Cope and Hardy wrote,
“It seems probable that this new rearrangement is similar in type to the Claisen rearrangement of allyl enol, phenol, and vinyl ethers and that it is motivated by similar forces.”48
Paper I of Cope's series The Rearrangement of Allyl Groups in Three-Carbon Systems, by Cope and Kathryn E. Hoyle and Dorothea Heyl,50 also at Bryn Mawr College, was published 17 months after the Cope and Hardy paper.48 In that paper, Cope et al. examined eq. 7 substrates with a wider series of substituents. They summarized the results of Paper I with eq. 8. Paper II in the series by Cope, Corris Hofmann, and Hardy examined several 1,5-dienes substituted to distinguish between what Cope et al. termed a rearrangement with inversion (from 3 to form 4) or a rearrangement without inversion (to form 5) (eq. 9). They observed only 4. The chemistry is complicated, in that there are two [1,3] rearrangements, not including possible (E,Z)-stereoisomers. Additional research in this area continued in Cope's laboratory for several years.50-55
In the 1940s and 1950s, Cope and his students performed extensive studies on what Woodward and Hoffmann later would term electrocyclizations.5 Ironically, Cope first studied sigmatropic rearrangements and then electrocyclizations. Woodward and Hoffmann did the reverse, in large measure because the theory for electrocyclizations is much less complex than is the theory for sigmatropic rearrangements. This is discussed in detail in Papers 1156 and 1257 in this series.
Cope's interest in medium sized-ring chemistry likely drew him into cyclooctatetraene (COT) chemistry, a topic of growing interest immediately after World War II. Walter Reppe's publication of the large scale, efficient preparation of COT via the tetramerization of acetylene was not published until 1948, but word had spread about this reaction after the interrogation of German scientists by the American occupying force.58 COT was first prepared by Richard Willstätter in 1911 in a series of steps including multiple (August Wilhelm von) Hofmann eliminations of trimethylammonium-substituted cyclooctanes.59
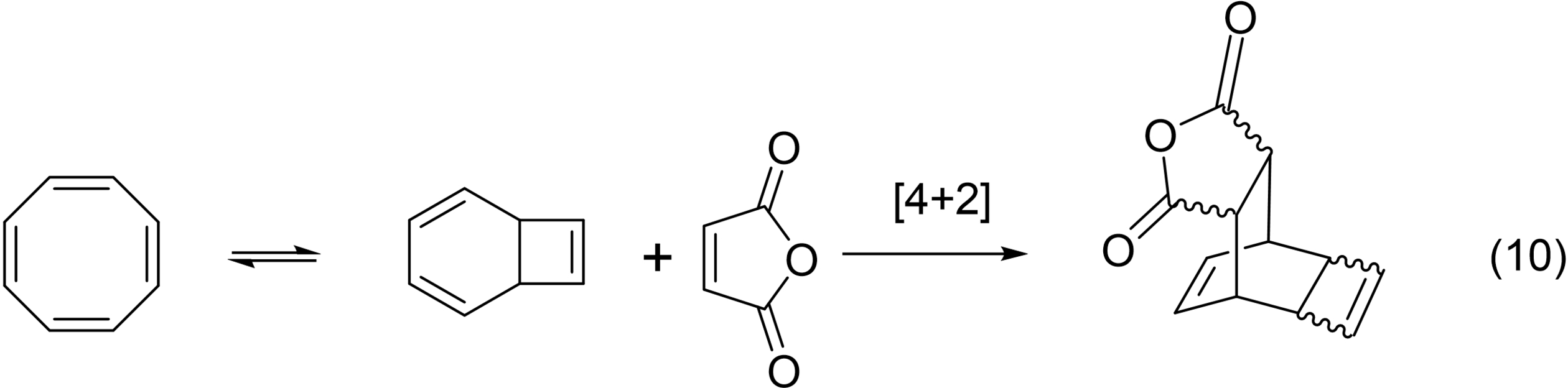
In Cope's first paper on medium-sized rings, he and his student Charles Overberger repeated Willstätter's 1901 synthesis and converted COT to its maleic anhydride (MA) Diels-Alder adduct (eq. 10). In both their 1947 communication60 and their 1948 full paper,61 Cope and Overberger simply referred to the product of COT and MA as an “adduct” without specifying its structure. In their 1947 paper, Cope and Overberger also reported that the mixed melting point of this adduct was “not depressed by mixture with a sample prepared from acetylene” without citing the source of the acetylene procedure. In 1948, Cope and Overberger cited the source of the COT as “prepared catalytically from acetylene” as the “FIAT Final Report No. 720, 1946, p. 26 (distributed by the Office of the Publication Board, U. S. Department of Commerce,” where FIAT was the abbreviation for “Field Information Agency, Technical.” Also in 1948, in a 92-page paper, the German chemists Reppe et al. revealed their nickel catalytic tetramerization of acetylene to COT.58 They also reported that the Diels-Alder adduct was not that from COT directly but rather from the bicyclo[4.2.0]octatriene (eq. 10). Thus, eq. 10 reveals two pericyclic reactions: the electrocyclization step followed by the cycloaddition step.
In 1950 and 1952, Cope et al. reported62 that 1,3,5-cyclooctatriene converts reversibly to an isomer that upon hydrogenation yields bicyclo[4.2.0]octane (eq. 11).63 Thus, Cope discovered another early example of an electrocyclization. Cope was able to separate 1,3,5-cyclooctatriene (6) from bicyclo[4.2.0]octa–2,4-diene (7) and each, when heated to 100 °C converted to the same equilibrium mixture. However, while 1,3,5-cyclooctatriene was inert to MA at 10 °C, bicyclo[4.2.0]octa–2,4-diene underwent smooth Diels-Alder reaction with MA.
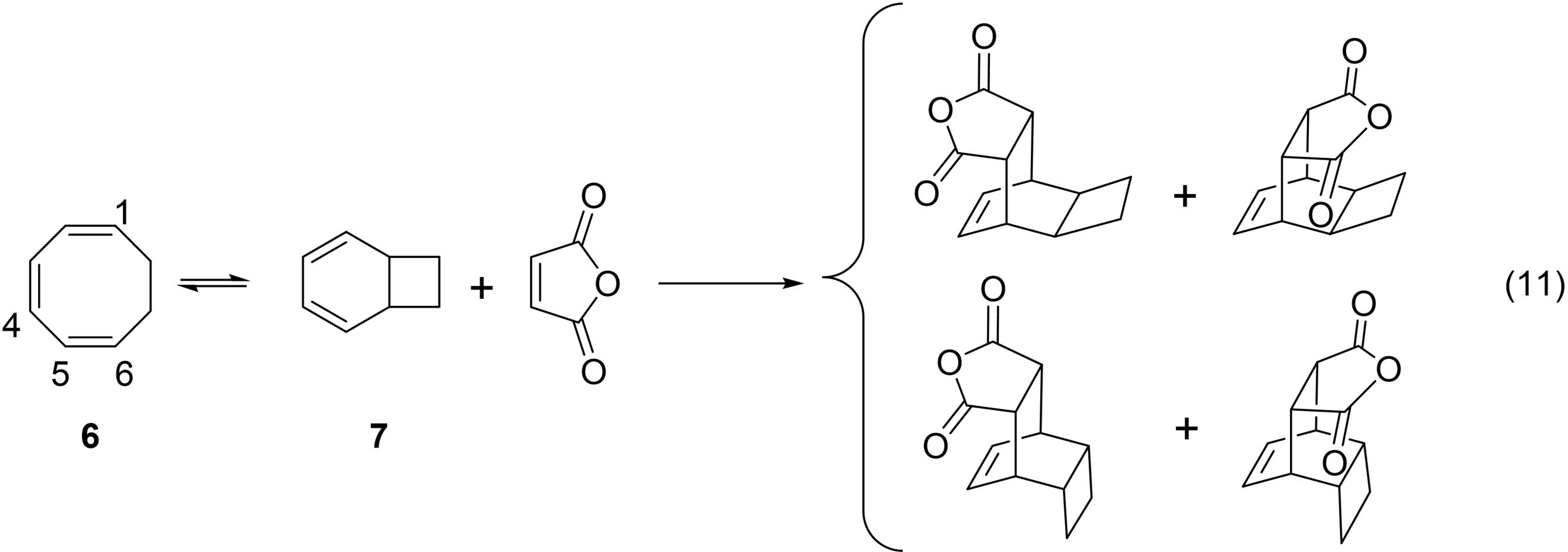
Of particular note, Cope observed that
“In the conversion of [6] to [7],… [eq. 11] the reaction is formally equivalent to an intramolecular Diels-Alder addition, in which C1 of [6] adds to C6, and C4 adds to C5 (forming a double bond).”62
This observation is identical to Woodward's observation in case of the resonance forms of benzene. In Scheme 1, the top equation is the normal representation of the two major resonance forms of benzene. Now, considering the bottom equation in Scheme 1, consider that the bonds in red do not exist. Then, the conversion of the left-hand graphic to the right-hand graphic is essentially a Diels-Alder reaction, just as imagined by Cope in 1952. As Woodward recalled in his 1973 Cope Award address, speaking of himself at age 12,

Illustration of Woodward's independent creation as a youth that the conversion of one resonance hybrid of benzene to the other can be viewed as a Diels-Alder reaction.64
“I recognized that if one were to assume the separate existence – however fleeting – of [the two resonance forms of benzene], the conversion of the one into the other might be regarded as a chemical reaction, in fact, an addition reaction of an olefin to a diene.”64
Also in 1952, Cope was the first to observe the thermal – actually, in this particular reaction, spontaneous – ring cleavage of a 1,3,5-cyclooctatriene to a 1,3,5,7-octatetraene, a valence isomerization reported frequently in the early 1960s (eq. 12).65-67
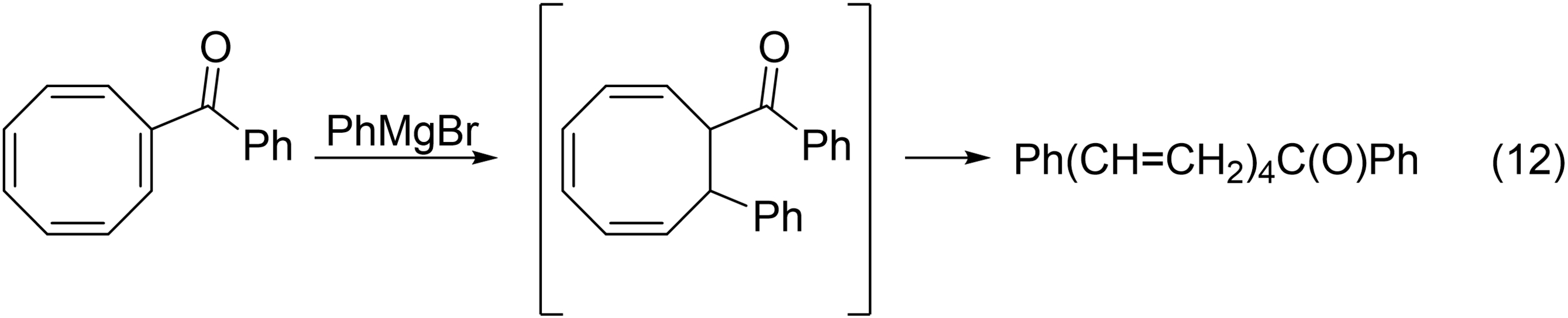
Cope's research group thus witnessed electrocyclizations, cycloadditions, and sigmatropic rearrangements in the 1940s and 1960s. How very close Arthur C. Cope was to discovering the Woodward-Hoffmann rules!
3.3.3. Why and Why Not Cope?
Why Cope? Because he was the inventor of the Cope reaction. Indeed, perhaps Cope's story belongs better in Paper 6 of this series4 with the other organic chemists in whose laboratory some of the key clues to the pericyclic no-mechanism problem were found.
Why not Cope? The W−H roadblock was twofold for Cope. First, there were few stereochemical hints in Cope's reactions. Second, he was little interested in photochemical reactions or aromaticity or in the mechanism of other valence isomerizations. And perhaps most importantly, Cope's interests lay elsewhere. In the last decade of his life, Cope studied the reactivity of medium-sized rings and what he termed “proximity effects” and “transannular reactions,” molecular asymmetry of olefins, e. g., the preparation of trans-cyclooctenes, and various aspects of molecular asymmetry. Perhaps Cope was also unaware of the no-mechanism problem, e. g., Emanuel Vogel's and Rudolf Criegee's cyclobutene ring opening mysteries (eq. 5) and Egbert Havinga's vitamin D problems (e. g., eq. 1 and eq. 2). Cope likely had little knowledge of molecular orbital theory or quantum chemistry, as these subjects do not seem to appear in his research. Even if Cope had an inkling, according to Jack Roberts and John Sheehan's description of him, an inkling would never have made it in print:
“One hallmark of Cope's research was meticulousness born of an almost morbid fear that some erroneous experimental result would be published under his name. At least once, this fear cost him priority for an important scientific discovery.”42
For the Woodward-Hoffmann story, Cope's chemistry was seminal for others such as Criegee, Vogel and Rolf Huisgen, who worked in the area. It was they, not Cope, who were close to Woodward-Hoffmann. But it was Cope's chemistry that led others to their foundational research as precursors to orbital symmetry.
3.4. Paul D. Bartlett (1907–1997)68, 69
3.4.1. General Background Information
Paul Bartlett received his B.A. from Amherst College in 1928. He studied with James Bryant Conant for his Ph.D., then worked at the Rockefeller Institute for a year and was on the staff at the University of Minnesota for two years. He then returned to Harvard where he remained until his retirement in 1972, at which time he became a Welch Professor at Texas Christian University until his second and final retirement in 1985.
In the 1940s to the 1970s, Bartlett (Figure 9) was one of the premier physical organic chemists in the world. He is primarily remembered for his work on bridgehead reactivity, hydride transfer from alkanes to carbocations, addition and chain transfer in polymerization, and photooxidations, elemental sulfur, and the mechanisms of solvolyses, including studies on the nonclassical carbocation.68-70

Paul Bartlett (left) with John D. Roberts, playing croquet, Colby College, 1948. Note the ties and white shirts, normal attire at Gordon Conferences in that era. Photograph courtesy J. D. Roberts.
3.4.2. Gateway: Pericyclic Reactions
In 1964, Bartlett and his students studied the thermal cycloadditions of highly halogenated olefins with several 1,3-dienes.71-73 In all cases, [2+2] products were formed and in several instances, [4+2] products were also obtained. Biradical intermediates were proposed as the first step in the proposed two-step cycloadditions. In 1965, Nicholas Turro and Bartlett studied the photosensitized cycloaddition reactions of haloethylenes and 1,3-dienes.74 The predominant reaction products were the [2+2] cycloaddition adducts formed via the triplet state of the diene on an unactivated olefin. Bartlett would continue his interest in these non-concerted cycloadditions in subsequent years, including writing reviews in Science in 196875 and in Pure and Applied Chemistry in 1971,76 the latter as a result of a invited lecture at an IUPAC meeting.
Bartlett's scientific meeting notes from the 1950s and 1960s have been saved with many of his papers at the Science History Institute. Figure 10 includes excerpts from four pages from these small spiral notebooks. Bartlett recorded the Vogel39 and Criegee16 cyclobutene→(E,Z)-1,3-butadiene conversions (eq. 5) from three meetings, in 1957,77 196278 and 1964.79 This transformation was one of Woodward's mysterious reactions64, 80 and served as a key hint that led to the Woodward-Hoffmann rules. Bartlett also attended – indeed, he was one of the presenters at – the 1964 Reaction Mechanisms Conference in Corvallis, OR where he lectured on Free Radical Reactions. Doering presented his lecture on Valence Tautomerism and No-Mechanism Reactions, where the famous Vogel and Criegee reactions were discussed.81, 82

Excerpts from three pages of meeting notes of Paul D. Bartlett. (A) Europe, 1957.77 (B) Germany, 1962.78 (C) From R. Criegee's lecture, Natick Conference, October 1964.83 (D) From Saul Winstein's lecture, Natick Conference, October 1964.83 Note in particular that he had written down the cis-3,4-disubstituted cyclobutene valence isomerization to the less stable butadiene product in (A), (B), and (C).
I have not found any indication that Bartlett was particularly interested in quantum chemical theory, molecular orbitals, or computational chemistry. I add one caveat to this statement. Bartlett did invite Roald Hoffmann to lecture at his and Frank Westheimer's combined group meeting on November 22, 1963.84 It was during the time period that Hoffmann visited Westheimer, E. J. Corey, Woodward and Bartlett, bringing with him a preprint of the first extended Hückel calculation focused on organic molecules.85 As Hoffmann recalled,
“November 22, 1963, was not just any day. It was the day of the Kennedy assassination. I remember a discussion whether to give the seminar or not. They decided to go on, but no one was listening!”86 [Figure 11]

Notes found in R. B. Woodward's archives written on November 22, 1963.87 (Left) “2:00 pm Your radio – I just heard that Kennedy (& Connolly) shot in car in Dallas.” (Right) “RADIO President Kennedy Shot from ambush – critically wounded.”
3.4.3. Why and Why Not Bartlett?
Why Bartlett? Because he was one of the foremost physical organic chemists of the day. Because he had studied 2+2 cycloadditions and understood the difference between them and 4+2 cycloadditions. He was also aware of Vogel's and Criegee's cyclobutene reactions (eq. 5 in Figure 1).39
Why not Bartlett? How close was Bartlett to solving the pericyclic no-mechanism problem?
In 1961 and 1962, of Bartlett's 11 scientific publications, six were on oxidation chemistry, two were on solvolysis mechanisms, two were on elemental sulfur, and one was on a miscellaneous topic. In 1964, all four of his scientific publications involved [2+2] cycloadditions. In 1965, five were on solvolyses, two were procedures for Organic Synthesis, two were on oxidation chemistry, one was on a miscellaneous topic, and one was on [2+2] cycloadditions. Thus, Bartlett had a sudden and somewhat abbreviated interest in cycloadditions in the early to mid-1960s, but these were of substates with highly polarized substituents.
That being said, Bartlett was well aware of these key clues to the pericyclic no-mechanism problem. Why was it not Bartlett instead of Woodward and Hoffmann?
Several factors can be proposed. Bartlett had little if any experience with molecular orbital theory. He was focused on non-concerted cycloadditions, not concerted reactions. He may well not have been familiar with the vitamin D chemistry as his research experiences were far distant from steroid chemistry. And he had his plate full, with oxidation chemistry and a solid interest in solvolysis mechanisms, nonclassical carbocation, and related chemistry also during the late 1950s and early 1960s.
Bartlett likely can serve as an excellent example of one of the leading practitioners of physical organic chemistry in the 1960s whose research interests were so distant from the pericyclic no-mechanism problem that none of them was in the race to its mechanistic solution.
3.5. Hyp J. Dauben, Jr. (1915–1969)
3.5.1. General Background Information
There were two Dauben brothers, both of whom were undergraduates at Ohio State University, both of whom did undergraduate research with William Lloyd Evans at Ohio State and published with him in the area of carbohydrate chemistry,88, 89 both of whom received their Ph.D. degree as organic chemists at Harvard University, and both of whom became academics at major research universities on the west coast of the United States. Hyp Dauben (Figure 12) studied with the physical organic chemist Paul D. Bartlett (see Section 3.4), then joined the faculty at the University of Washington in 1945. His brother William G. Dauben studied with Patrick Linstead and Louis F. Fieser, then joined the faculty at the University of California, Berkeley in 1945.90 Both could have solved the pericyclic no-mechanism problem. Here is Hyp Dauben's story. William G. Dauben's story appears in Paper 6 in this series.4

Hyp J. Dauben in his office at the University of Washington, 1969. Photograph courtesy John D. Roberts.
In contrast with his brother, Hyp Dauben was not a highly published academic. According to SciFinder Scholar, he published only 42 scientific publications in his career, five of which were from his graduate school days.
3.5.2. Gateway: Aromaticity
Of relevance to the no-mechanism problem is Dauben's interest in aromaticity. The first two communications in the February 1951 issue of the Journal of the American Chemical Society were groundbreaking. Two research teams, Dauben and Howard J. Ringold in Washington91 as well as Doering and Detert at the Hickrill Chemical Research Laboratory in Katohah, NY92, 93 reported the synthesis of tropone and provided strong evidence of its aromatic character. Doering and Detert also made the first formalism of what has been called Hückel's “4n+2” rule. Actually, Doering and Detert wrote “2+4n.” Dauben and Ringold suggested that tropone's aromatic character
“may originate from carbonyl polarization and resonance of six π-electrons among seven π-orbitals. This system would acquire a benzene-like 2pπ molecular orbital energy pattern. . . ”91
In 1957, Dauben reported the synthesis of various cycloheptatrienylium salts (eq. 13).94 In 1958, he reported on ion π-complexes of tropenium salts.95 And in 1961, Dauben published a paper on the heptalenium ion.96
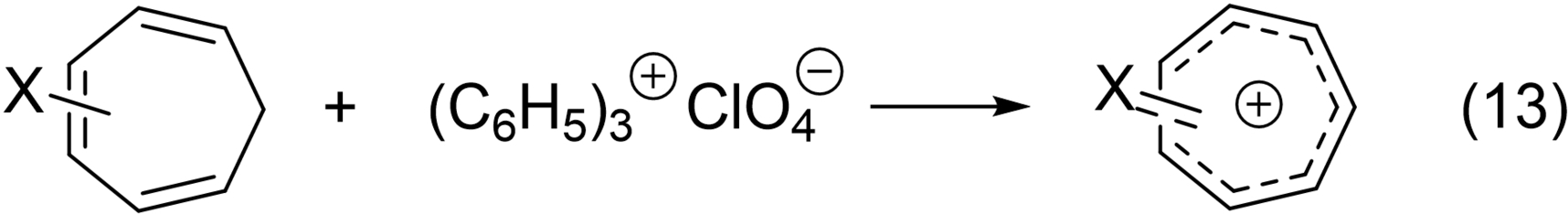
In 1963, Dauben published a paper on Cycloheptatrienide (Tropenide) Anion, a “4n” monocyclic” compound, “as solutions or suspensions of its [potassium] salt.”97 Based on acidity values, Dauben reported,
“it may be approximated that the empirical resonance energy of the tropenide anion is about 0.8–1.1β greater than that for cycloheptatriene.”97
Dauben then was in the midst of discussions of aromaticity, a concept which did not lead him into antiaromaticity but was, just two years later, done so by Breslow and Dewar.98-100
3.5.3. Why and Why Not Hyp Dauben?
Why Hyp Dauben? Because he was an expert in aromaticity. He could have imagined the concept that was used several years later when Zimmerman101, 102 and Dewar103, 104 introduced Hückel and Möbius/aromatic and antiaromatic transition states for pericyclic reactions.
Why did Hyp Dauben not discover the solution to the pericyclic no-mechanism problem? He was primarily focused on aromaticity and preparing a variety of nonbenzenoid aromatic compounds. He was not into valence isomerizations nor the no-mechanism problem. He had not discovered antiaromaticity that awaited Breslow98, 100 and Dewar99 in 1965.
Interestingly, Dauben was the Sloan Visiting Professor fall term 1964 at Harvard. In principle, he could have contributed to the extension of the W−H rules from electrocyclizations to cycloadditions and sigmatropic rearrangements at that time. Was he aware of Hoffmann's research? Probably not, as Hoffmann had temporarily shelved his collaborative research with Woodward from late June to mid-October 1964.56 On the other hand, had Dauben talked with Subramania Ranganathan about his research,105 Dauben might have become interested in the pericyclic no-mechanism reaction. But that did not happen.
3.6. Derek H. R. Barton (1918–1998)106-111
3.6.1. General Background Information
In 1950, Derek Barton (Figure 13) published “The Conformation of the Steroid Nucleus”26 in the journal Experientia, which transformed organic chemistry and led to his receipt of the Nobel Prize in 1969. Barton's highly productive career ended only with his death in 1998, just hours after a full day with his research group at Texas A&M University in College Station, Texas. Barton was also a well-travelled chemist, not just on the lecture circuit but in his various academic positions. In 1957, he moved from Glasgow to assume the professorship in organic chemistry at Imperial College, a position he maintained until his forced retirement 20 years later. He then moved to France and the CNRS. the French National Centre for Scientific Research, until he was forced to retire a second time. Barton then moved to Texas A&M where he essentially died at the bench.109-113 He never really did retire.

Derek H. R. Barton, Birkbeck College, London, 1954. Photograph courtesy D. H. R. Barton.
Barton was a very active chemist! Between 1960 and 1965, according to SciFinder, the years just before W−H, Barton published well over 100 papers in the fields of natural products structure identification, synthesis, the development of novel synthetic methods, photochemistry of terpenoids, and biosynthesis. If it was a terpenoid, Barton was interested in it. That especially included steroids.
3.6.2. Gateway: Pericyclic Reactions
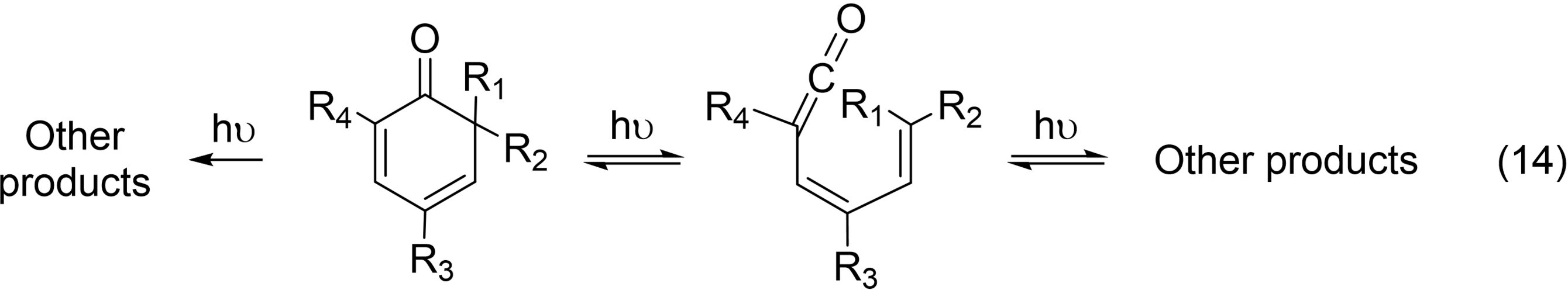
In 1959 and 1960, Barton and Gerhard Quinkert114, 115 reported on the very easy photochemical valence isomerizations shown in eq. 14.
Also in 1959, Barton published a singly-authored perspective – not a review paper – entitled “Some Photochemical Rearrangements”116 based on his lecture given on June 16, 1959 on the occasion of his receipt of the first Roger Adams Medal. The Roger Adams Medal, together with the Arthur C. Cope Award, are the premier awards in organic chemistry bestowed by the American Chemical Society. Why this paper was published in the Swiss journal Helvetica Chimica Acta rather than an ACS journal may reflect that, at that time, the ACS did not have a journal that published perspectives and accounts of individual's chemical research.
In any event, in that 1959 paper, Barton made several far-reaching hypotheses. He wrote,
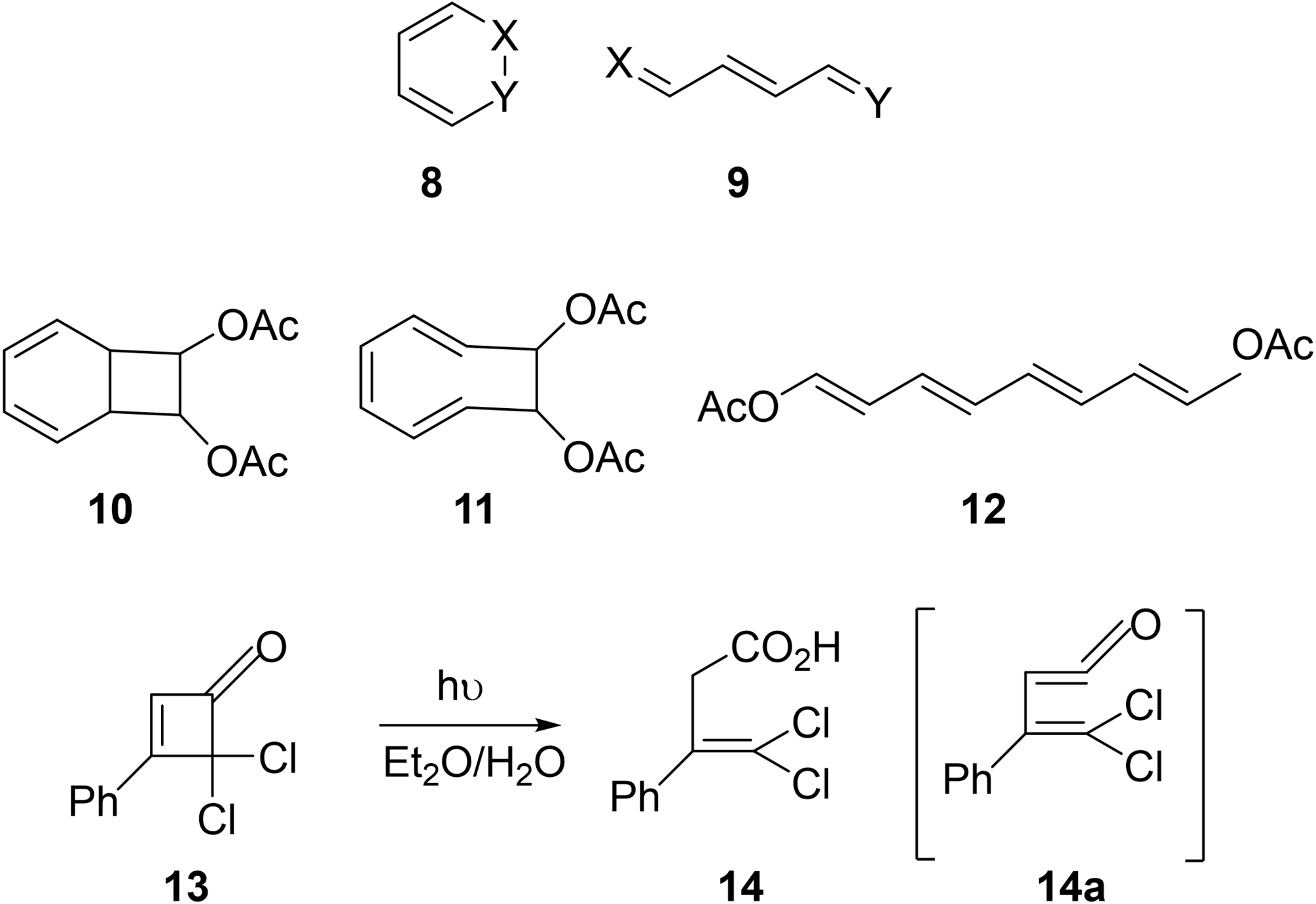
“Ring cleavage of the type [8] → [9] should be a general reaction. It is not, of course, required that methine groups (CH) be present; they could well be replicable by the appropriate hetero-atoms or other groupings. This generalization, which should be of value in predicting further photochemical reactions, can be extended as follows. If one considers any ring of 2n members containing (n-1) conjugated double bonds then, in principle, irradiation… should furnish an open chain compound containing (n-1+1)=n conjugated double bonds. In support of this idea, we have been able to show that irradiation of [10] in ether produces, via [11], the tetraene [12]. Although we have not yet obtained the latter in a state of purity, it shows the correct ultraviolet maxima… Similarly, irradiation of the cyclobutenone [13] gave [14] presumably via 14 a]…”116
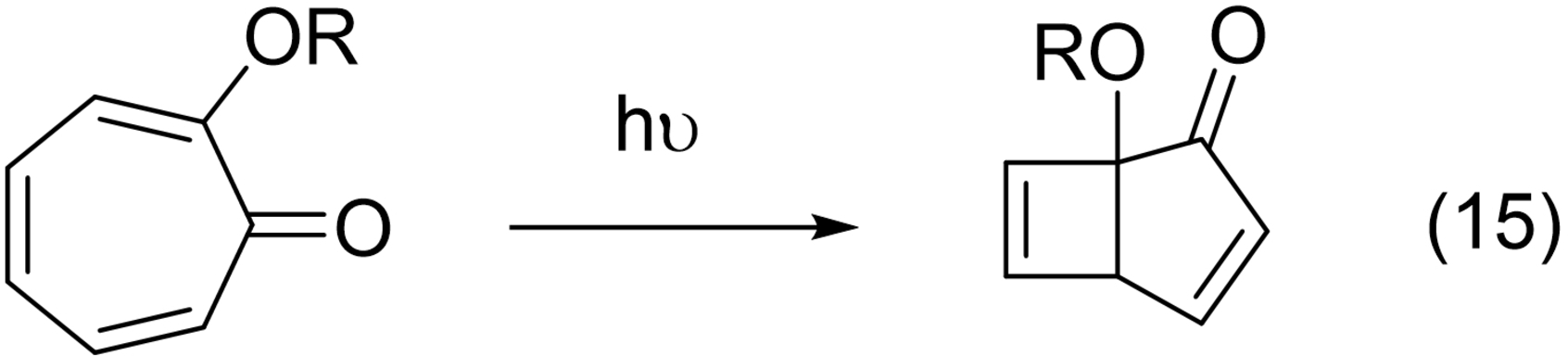
“Quite different considerations apply to rings that contain (2n+1) members and n conjugated double bonds. Here it can be predicted that cleavage reactions should not be observed, only bridging reactions. This is certainly true for the seven-membered tropolone ring… ”116 (eq. 15)
In 1959, six years before Woodward and Hoffmann, Barton was on the right path toward organizing a wide body of valence isomerization literature and developing a theory to explain the variety of reaction mechanisms. Barton had twice before developed a set of chemical rules: in the late 1940s and early 1950s, he proposed a rule that related the differences in molecular (optical) rotation to chemical structure, mostly for steroids.111 As Barton summarized,
“What was published was the integration of a large body of often confused and misleading literature. The interpretation required critical analysis and judgment.”111
The second set of chemical rules was that of conformational analysis, also based on his analysis of steroid structure and reactivity. As is well understood today but not in the early 1950s, Barton explained the stability and relative reactivity of axial and equatorial substituents on cyclohexane rings.26, 111
3.6.3. Why and Why Not Barton?
Why? First, because Barton had already experienced proposing a major concept that changed the face of chemistry, that of conformational analysis in the early 1960s.26, 27 Not all scientists have been intimate with that kind of experience, to know that a single person can make such a big difference. Perhaps that is why many folks choose smaller, more tightly defined problems to examine.
Barton also had a knowledge of the photochemistry in various pericyclic reactions.
Why not Barton? For Barton to propose a set of rules governing photochemical valence isomerizations – electrocyclizations – followed his pattern of integration of a large swath of the chemical literature, he would have had to include a broader set of chemical clues. What Barton did not include in his analysis were other experimental data, including Criegee's16 and Vogel's39 cyclobutene ring opening, the photochemical but not thermal concerted [2+2] cycloadditions, the stereospecificities within the vitamin D series – some of which were not known in 1959 but would be revealed within a year or two. It is possible that Barton had not followed small ring chemistry, given that his interests at the time were natural product chemistry, steroid chemistry, photochemistry, and structure identification.
Barton also displayed no knowledge of molecular orbital theory. Had he known of the pericyclic no-mechanism problem, his lack of MO theory would have rendered that knowledge moot – unless he consulted with a theoretician or physical chemist.
And thus, Barton was close but not close enough.
3.7. Sara Jane Rhoads (1920–1993) and Paul de Mayo (1924–1994)
3.7.1. Background and Gateway: Sigmatropic Rearrangements
Sara Jane Rhoads received her Ph.D. from Columbia University in 1949. Her Ph.D. advisor was William von Eggers Doering with whom she published on 2- and 3-Vinylpyridines in the Diels-Alder Reaction117 and on the alkylation of β-keto esters.118 She taught at the University of Wyoming from 1948 until her retirement in 1984 (Figure 14).

Sara Jane Rhoads, ca. 1980. Photograph courtesy Edward L. Clennan.
Rhoads is mostly known for her teaching, especially with undergraduates. She initiated the undergraduate research program at Wyoming. Her few publications were centered around what later became known as sigmatropic rearrangements. No doubt this interest stemmed from her graduate school years with Doering. In the 1950s, she published five papers on the Claisen rearrangement (eq. 6 in Figure 1).119-123 In the early 1960s, her attention reverted to the chemistry of β-keto esters, one of her thesis projects. On May 24, 1962, Rhoads applied for a sabbatical leave with John D. Roberts at Caltech, explaining to Roberts that she wanted
“to gain some first-hand knowledge of NMR techniques and applications and would like very much to do this at Cal Tech if you would be willing and able to accept me in your group for a few weeks. If you offer a formal course or ‘orientation’ program in this area I would try to arrange my time in California to coincide with these as closely as possible… we have been interested in β-keto esters of the medium-sized rings for the past few years. I believe that NMR studies of these systems might reveal some interesting things about the enolic forms. So, if I may, I would like to bring a few along to see if such a study is worth pursuing.”124
Rhoads's short sabbatical with Roberts was successful in that in the next few years, she published four papers on β-keto esters.125-128 But did Rhoads learn any molecular orbital theory from Roberts who had just completed his book Notes on Molecular Orbital Calculations129 and who was including MO theory in many of his research publications130-133 and his lectures and organic chemistry courses?134, 135 There is no evidence that MO theory became important to Rhoads.
In 1963, and of relevance to the Woodward-Hoffmann rules, Rhoads published a chapter in Paul de Mayo's (Figure 15) two volume set Molecular Rearrangements entitled Rearrangements Proceeding Through ‘No Mechanism’ Pathways: The Claisen, Cope, and Related Rearrangements136 This chapter was surely stimulated by her research on the Claisen rearrangement and her graduate studentship with Doering, the inventor of the term “no-mechanism.”137, 138 In the introduction of her chapter, Rhoads defined a no-mechanism reaction (definition reproduced in the introductory section of this paper). Contained within Rhoads's chapter are examples of electrocyclic reactions, cycloadditions, and sigmatropic reactions.

Paul de Mayo received his Ph.D. with Derek Barton and had a postdoctoral fellowship with R. B. Woodward. de Mayo had a brilliant career as a natural products chemist and photochemist139 but perhaps his most singular achievement was the two-volume set on Molecular Rearrangements published in 1963140 and 1964.141 The first volume included Sara Jane Rhoads's chapter on the no-mechanism reaction.136 Photograph courtesy Myra Gordon and Kim Baines, University of Western Ontario.
Figure 16 illustrates three reactions with transition states from Rhoads's chapter. Rhoads describes transition state (Figure 16, reaction A) as “part of an aromatic system”136 while transitions states (reaction B) and (reaction C) are illustrated as having aromatic-like, delocalized transition states.136 In this sense, Rhoads was a precursor to Dewar's 1966 characterization of pericyclic reactions having aromatic-like (or antiaromatic) transition states.103, 104, 142

Three graphical excerpts from Rhoads's 1963 chapter on no-mechanism reactions.136 Regarding reaction B, Rhoads pointed out that the stereochemistry of the intermediate triene(s) was not actually determined.
Rhoads also refers to the Havinga and Schlatmann paper in which the Oosterhoff orbital symmetry explanation is proposed.40
In 2013, Hoffmann described the importance of Rhoads's chapter to him in the 1960s,
“That review was a good source for the literature for me, as we began to understand. We can′t blame her too much, even people closer to orbital thinking than Sarah missed the clues out there.”143
3.7.2. Why and Why Not Rhoads?
Why Rhoads? She knew the no-mechanism problem intimately.
In 1965, Rhoads was certainly in position to have extended Oosterhoff's suggestion.40 She had read the Havinga paper in which it appeared; or at least, she had cited it.136 And she knew much of the chemistry. She certainly knew of the no-mechanism problem, having written an entire review article on the subject (though it did omit key reactions, like those of Vogel and Criegee (eq. 5)). She also had the opportunity to know of the MO explanation: In her 1963 review,136 Rhoads also had cited Havinga's 1961 publication40 in which appeared Luitzen Oosterhoff's orbital symmetry explanation for the vitamin D mechanistic anomalies.
Why not Rhoads? Perhaps she knew too little molecular orbital theory – possibly through her short sabbatical with Jack Roberts144 who was the organic chemist expert in MO theory. In the early 1960s, most organic chemists considered MO theory to be rather outside their radar screen. However, her interest with Roberts was NMR, not MO theory. After 1963, Rhoads published little. She continued to study thermal rearrangements,145 wrote a major review in 1975 on the Claisen and Cope rearrangements for Organic Reactions146 and published on C- versus O-alkylation in cyclic β-keto esters.128
Fundamentally, Rhoads was not in the mainstream of original research in chemistry. She was a substantial contributor and a substantial educator but in terms of a contributor to state-of-the-art chemistry, her involvement was in a much more limited sense.
3.8. Jack Hine (1923–1988)
3.8.1. Gateway Information
During the time-period discussed in this paper, many chemists were performing research that was related to what eventually became the Woodward-Hoffmann rules. As discussed above and in more detail in an earlier paper in this series, the use of molecular orbital theory was one absolute requirement in the W−H solution. One aspect of that theory was the recognition of the presence of phases/nodes in all but the lowest MO of every compound as well as in the overlap of neighboring orbitals or orbitals interacting in a reaction or charge-transfer complex. Frequently authors drew what they thought were the molecular orbitals relevant to a particular chemical situation but what they drew were either the basis orbitals or the first molecular orbital of the compound with zero nodes.
In a 1955 paper, Jack Hine et al. wrote the following about bicyclo[2.2.2]octatriene, a compound synthesized by Howard Zimmerman just five years later.147
“It may also be seen to be impossible to arrange the π-orbitals in a way which gives all overlapping lobes the same algebraic sign… Because this is the first case known to us in which the sign of an atomic orbital has been of such importance, bicyclo[2.2.2]octatriene is of particular interest.”148

In the second edition (1962) of his textbook Physical Organic Chemistry,148 Hine presented a short discussion on MO theory and included the molecular orbitals of 1,3-butadiene. And even though he cited Doering and Sara Jane Rhoads in his text, Hine did not discuss the no-mechanism problem. This may well be due to its date of preparation. While the book was published in 1962, there are no references to 1961 and very few to 1960. The no-mechanism term seems to have been first used in the chemical literature in 196265, 136, 138 though in conversation, surely earlier.149 Hine did discuss the Cope and Claisen rearrangements but did so in a chapter entitled Some Four-Center-Type Rearrangements,262 although he clearly draws a curly arrow mechanism involving six carbon atoms for these rearrangements.
3.8.2. Why and Why Not Hine?
Why Hine? Because he was a solid physical organic chemist with a knowledge and appreciation of molecular orbital theory, aromaticity, and reaction mechanisms.
Why not Hine? Why not anyone who knew the MOs of 1,3-butadiene? Did he know of or even care about the no-mechanism problem. One can only speculate. He, as many of his peer group of the time, may not even known of the pericyclic no-mechanism probem. Or considered the several examples of “laboratory curiosities”150 very important.
3.9. Ronald C. Breslow (1931–2017)151-153
3.9.1. General Background Information
Ronald Breslow received his A.B., M.A., and Ph.D. in 1955, all from Harvard University, ultimately studying with Woodward in graduate school. Oddly, while Breslow published two papers in 1953 as an undergraduate with Gilbert Stork,154, 155 he had no papers with Woodward. After a year as a National Research Council Fellow in Cambridge (England), Breslow joined the faculty of Columbia University where he remained for the rest of his life. Breslow was an active researcher until the day he died.
3.9.2. Gateway: Aromaticity
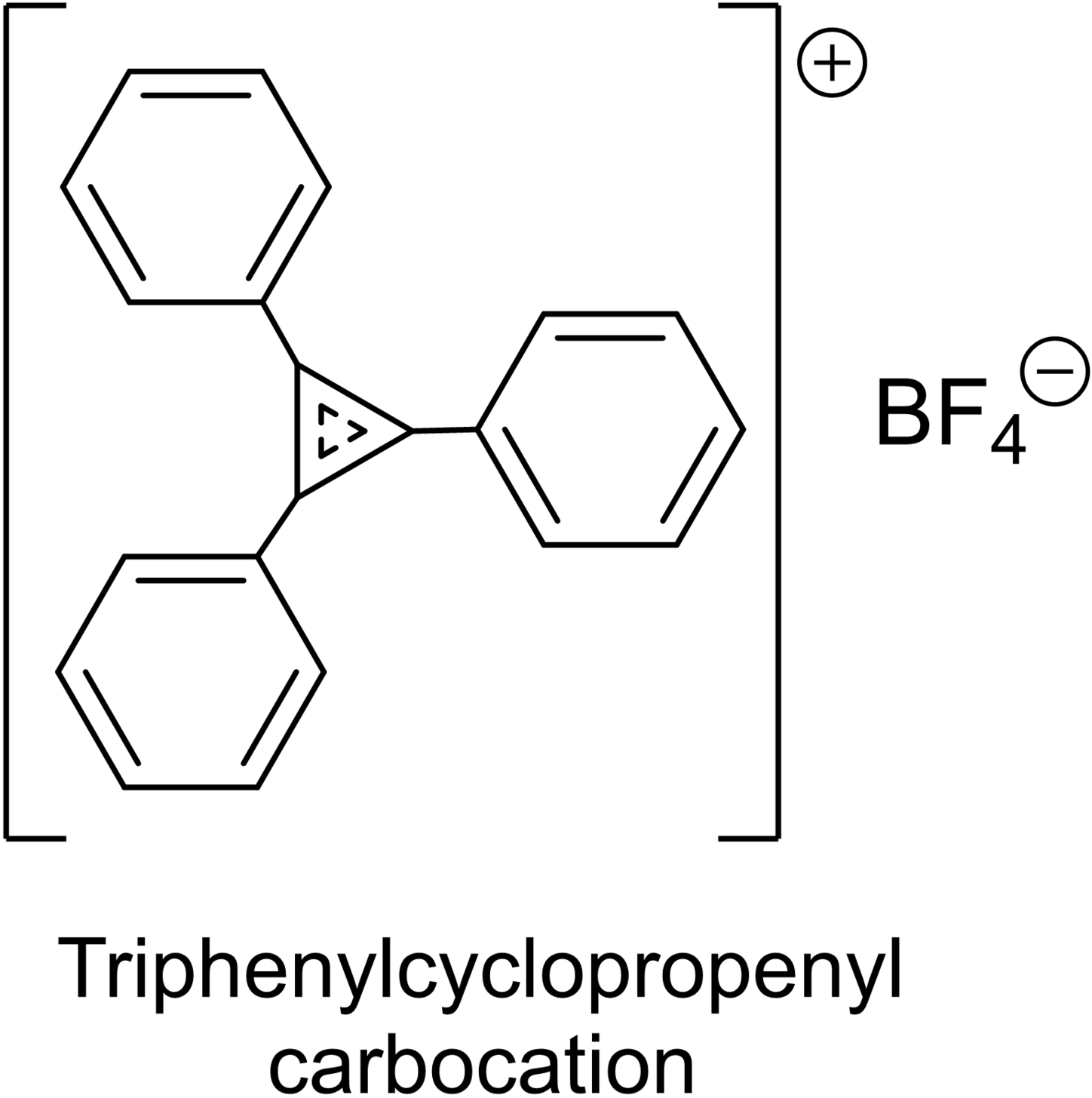
The most important experimental data pointing to the void that was solved by Woodward and Hoffmann – the solution to the pericyclic no-mechanism problem – was certainly the expanding collection of alternating reaction allowedness/forbiddenness and alternating reaction stereospecificities. Ronald Breslow (Figure 17) was the preeminent chemist who studied antiaromaticity. Indeed, it was Breslow who, in two publications in 1965,98, 100 one being a perspective published in Chemical & Engineering News,100 first coined the term antiaromaticity. (Michael Dewar, to be discussed below, simultaneously proposed that term.99) According to Hückel's 4n+2 rule of aromaticity, the simplest aromatic system would be the cyclopropenyl cation, with n=0. In 1958, Breslow – doing the chemistry with his own hands – synthesized triphenylcyclopropenyl carbocation156 and in 1967157 synthesized the unsubstituted cation itself.

Ron Breslow standing between Nick Turro (left) and Gilbert Stork, Columbia University, 1960s. Photograph courtesy R. Breslow.
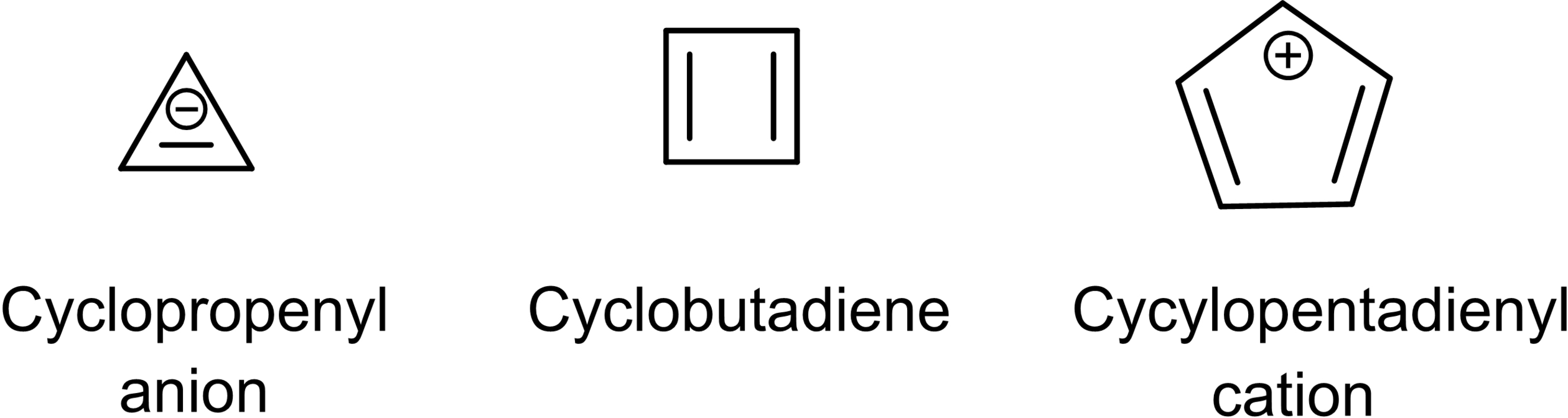
When Breslow began his independent academic career at Columbia University in 1956, it was not clear if Hückel's 4n compounds would be nonaromatic or particularly destabilized, i. e., antiaromatic. Breslow and his group concentrated their efforts on three 4 π species, namely the cyclopropenyl anion, cyclobutadiene, and the cyclopentadienyl cation, all of which are indeed antiaromatic.151-153, 158, 159
In the 1950s and 1960s, Breslow's aromaticity-imagination was unrestrained. Among the various systems he examined that might have aromatic properties were seven-membered rings having ten electrons (4n+2, n=2). One of these was 23, experimentally modelled by 24, was found not to be aromatic.160 In that same 1963 paper, Breslow reported the predicted Hückel molecular orbital calculated delocalization energies as a function of the number of π-electrons (Figure 18). Clearly there is alternation in delocalization energies following Hückel's 4n+2 rule.
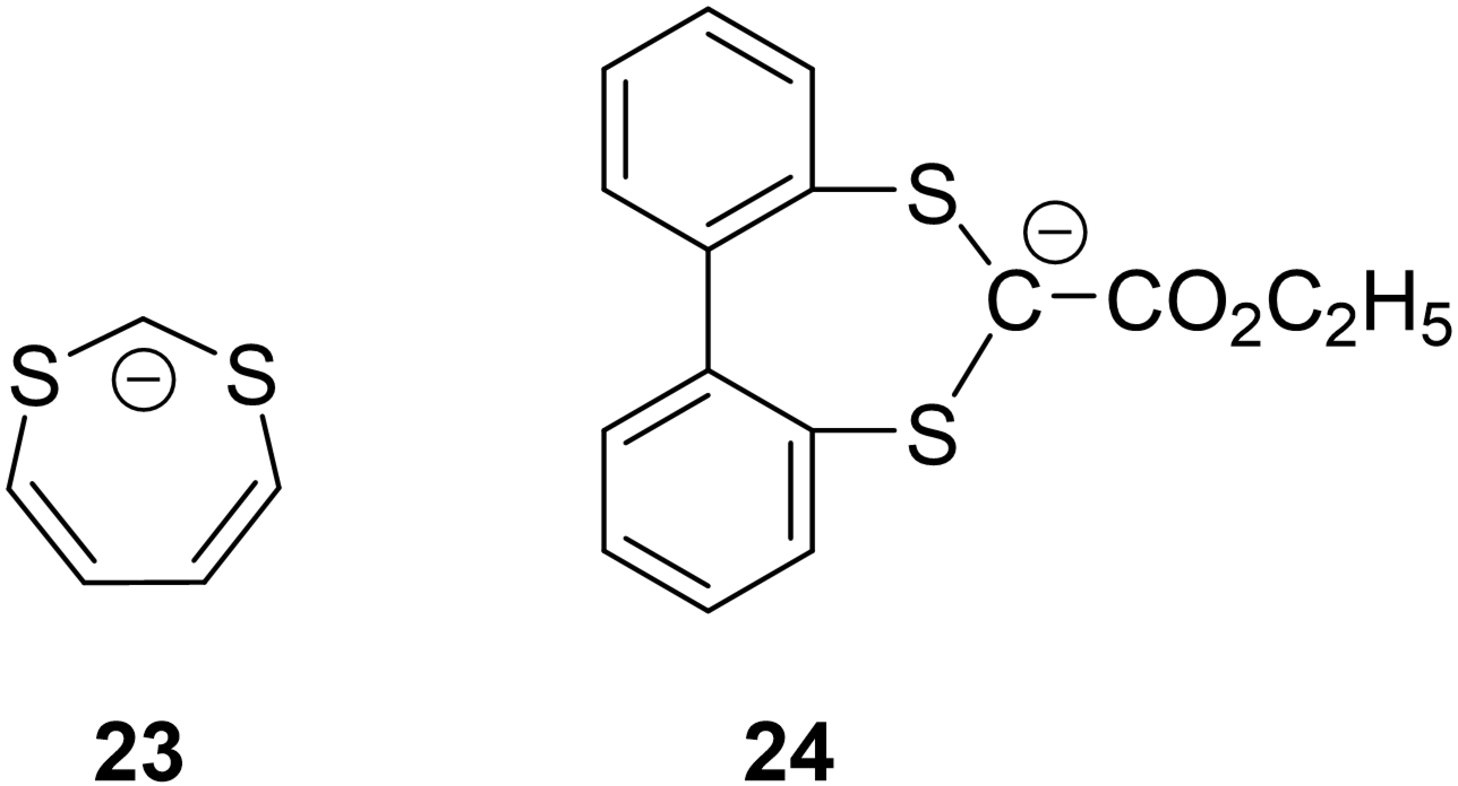

“Difference in π electron delocalization energies (from a Hückel calculation) between cyclic and linear simple conjugated systems as a function of the number of π electrons.”160
3.9.3. Why and Why Not Breslow?
Why? Because he had the most knowledge and understanding of the alternating aromatic-antiaromatic physical and chemical properties of molecules of all chemists in January 1965, when the first W−H paper was published.5 He also had experience with molecular orbital theory, given that he was the first,98, 100 along with Dewar,99 to discover and define antiaromaticity in quantum chemical terms. And, as a leading physical organic chemist who was studying the chemistry of small ring compounds, likely he was aware of the many valence isomerizations that were clues to the pericyclic no-mechanism problem.
Why not? As close as Breslow was to orbital symmetry, his mind was dominated by aromaticity and antiaromaticity. (That sounds like a paradox.) He was also familiar with MO calculations. However, Breslow was likely not aware of the other forms of alternation chemistry that were in the air at the same time, especially the vitamin D chemistry of Havinga and his group at the University of Leiden.40 So, he focused entirely on the aromaticity-antiaromaticity theme and missed the no-mechanism problem entirely. Indeed, even after Woodward and Hoffmann published their five communications in 1965 using frontier molecular orbital theory, correlation diagrams and perturbation theory, Breslow did not rush in as did Howard E. Zimmerman101, 102, 161 and Dewar103, 104 and propose the alternative explanation of aromatic versus antiaromatic transition states in pericyclic reactions.
3.10. Douglas E. Applequist (1930–2016)
3.10.1. Background and Applequist's Harvard Course
Douglas Applequist (Figure 19), a physical organic chemist, received his Ph.D. with John D. Roberts in the mid-1950s at Caltech. Applequist's expertise was small ring chemistry, a field that he studied with Roberts and that he continued in his not-extensive research at the University of Illinois. Applequist took his spring 1964 sabbatical at Harvard. During that semester, he taught a graduate level course on small ring chemistry. Among the attendees at that course was Roald Hoffmann who took voluminous notes which have been provided by Hoffmann to this author. Careful examination of the notes reveals the material presented by Applequist relevant to the history of the development of the Woodward-Hoffmann rules.

Douglas Applequist at the University of Illinois, Urbanna-Champaign, IL, ca. 1965. Photograph courtesy D. Applequist.
Mixed throughout the series of lectures are examples of all three major classes of reactions which would in 1969 be termed pericyclic reactions (electrocyclizations, cycloadditions, and sigmatropic rearrangements). This includes Diels-Alder reactions and several Cope reactions involving cyclopropyl rings. In contrast, there are only several very brief mentions of molecular orbital theory, and not a single example of showing the molecular orbitals of a molecule such as 1,3-butadiene. Rather, from the beginning, there is a discussion of hybridization, VB estimates of strain energies, and some estimates of strain energies using classical methods. Atomic orbitals are drawn in some cases, e. g., Walsh diagrams of cyclopropane.
As shown in Figure 20(A), Applequist discussed the cyclopropyl-X to allyl electrocyclization for the carbene, free radical and carbanion but not for the carbocation (or the solvolysis of cyclopropyl-L (L=leaving group) for which, by 1964, there was some examples of reaction stereospecificity that would be explicable only by the orbital symmetry rules. In Figure 20(B), Hoffmann recorded several examples of cyclobutene ring openings that are consistent with thermal conrotatory motion. Vogel's39 and Criegee's16, 39 stereospecific cyclobutene ring opening reactions (eq. 5) are shown. At the very bottom of Figure 20(B), in light font and very hard to read, Hoffmann noted that “trans starting material [cyclobutenes thermolyzes] faster.” One example on this page is the difficulty of ring opening of the cyclobuteno-dihydrophenanthrene.162 The results on this page are consistent with what Woodward and Hoffmann would shortly thereafter find: thermal conrotatory ring openings of cyclobutenes.5

Pages from Hoffmann's notes of Douglas Applequist's lecture notes, Harvard University, Spring 1964. (A) Page 58. (B) Page 87. Vogel's and Criegee's16, 39 stereospecific cyclobutene ring opening reactions are shown. At the very bottom, very hard to read, Hoffmann wrote for the 3,5-disubstituted cyclobutenes, “trans starting material [thermolyzes] faster.” Note the difficulty of ring opening of the cyclobutenyl derivative of dihydrophenanthrene. The results on this page are consistent with thermal conrotatory ring openings of cyclobutenes.163
Applequist's lectures included several key hints that could have led to the orbital symmetry rules including the use of the term “no-mechanism” (Figure 21) and other pericyclic reactions such as cycloadditions (also shown in Figure 21). But the problem was not identified as such by Applequist nor were the examples pulled together in a cluster. One false counter example (Adam's chemistry that contained some structural misassignments164) may have muddied the waters. Neither Hückel nor any molecular orbital theory is documented in Hoffmann's notes of Applequist's course. Ironically, sitting in the lectures was one of the developers of the extended Hückel method who could have and was soon to explain some of the no-mechanism stereospecificities, that of course being Hoffmann.

Page 83 from Hoffmann's notes of Douglas Applequist's lecture notes, Harvard University, Spring 1964. Hoffmann writes “Cycloadditions by constrained enophiles” and of the “4 center, no mechanism” reaction.”163
To the extent that Hoffmann's notes represent Applequist's lectures, and given the detail of the notes, one might ask: What is the relevance of these notes to the history of the development of the W−H rules?
-
The notes illuminate Applequist's knowledge and suggest that he was in a prime position to solve the pericyclic no-mechanism problem.
-
The notes reveal what Applequist's students learned in his course, indicative of their position to solve the problem.
-
With the reasonable conclusion that Applequist's knowledge was not uniquely better than that of other physical organic chemists of his peer group around the world, the notes suggest that others were also in the position to solve the problem.
-
And on the reasonable assumption that other chemistry faculty taught a course similar to that of Applequist's, then graduate students, postdoctorals and others around the world were taught the same material. For example, Berson and Zimmerman team-taught such a course at the University of Wisconsin, as did Hyp Dauben at the University of Washington. Roald Hoffmann calls himself “lucky”86 that he was able to take Applequist's course. Other organic chemistry students at Harvard in the spring of 1964 did not do so, perhaps to their disadvantage. Their lack of attendance is an example of the tension between time available and choices people make regarding their time.
3.10.2. Why and Why Not Applequist?
Why? Applequist was a leading physical organic chemistry with research and teaching interests in small ring chemistry. Hoffmann's notes of Applequist's lectures at Harvard University revealed his knowledge of many of the pericyclic no-mechanism anomalies.
Why not? In spite of his knowledge of these anomalies, Applequist did not focus on the no-mechanism problem nor apparently see it in its entirety. Perhaps he was not aware of the steroid examples in the vitamin D series, for example.40, 165 Or Corey and Hortmann's dihydrocostunolide thermal and photochemistry.166, 167 And perhaps he was not convinced of the value of molecular orbital theory which was necessary to solve the pericyclic no-mechanism problem (see below). As Hoffmann summarizes,
There is mention of MOs in Applequist's lecture, but most discussion in terms of valence bond. First few pages, discussion of hybridization, VB estimates of strain energies, no MOs. There was a chance of MOs but it is not there. That MOs had not penetrated to this person teaching a progressive course in physical organic chemistry suggested to me maybe that there is a place for me for MO calculations. The two-hour exams do not ask anything about orbitals.”168
As Applequist himself recalled in a 2004 letter to Hoffmann,
“The problem is that at that period of time [late 1950s to 1965], I did not even begin to appreciate the importance of what was being created under my nose. I was used to the fashion among organic chemists to embellish their publications with molecular orbitals and especially frontier orbitals, but I saw very little there of predictive value beyond the 4n+2 rule for aromaticity. My own Ph.D. thesis was a test of some LCAO calculations by Jack Roberts on methylenecyclobutene, and the calculations proved misleading.”169
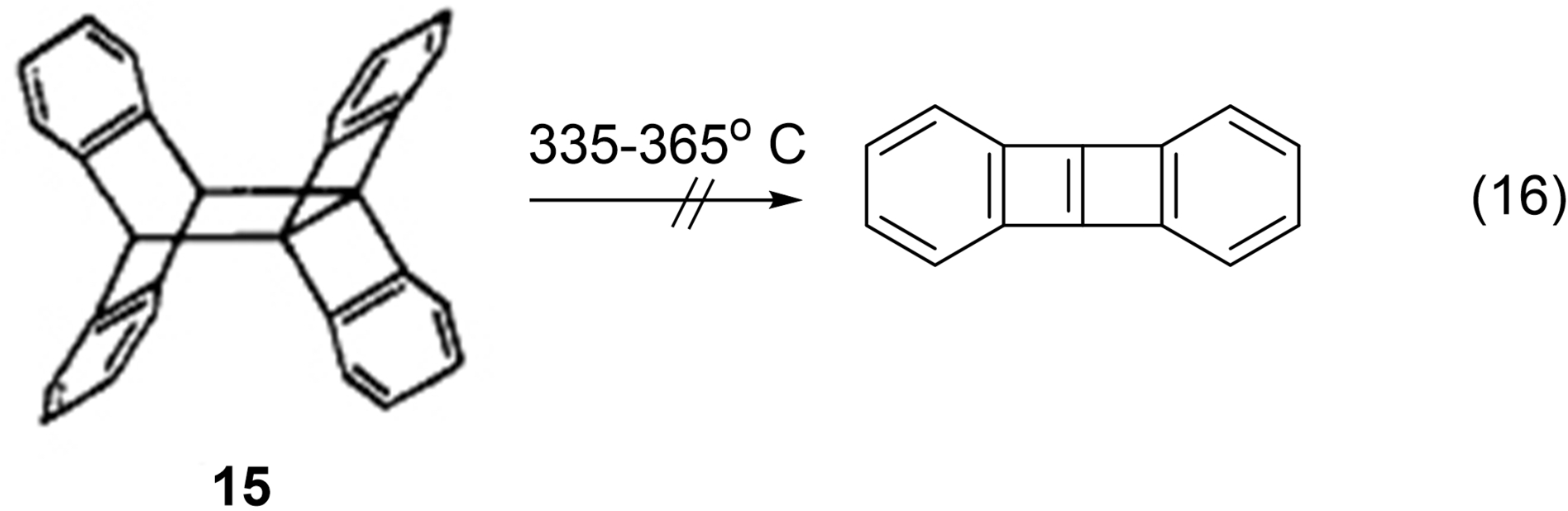
There was another reason for not discovering the orbital symmetry rules, like all chemists described herein. Applequist, like all the others, was busy in other research programs and other educational responsibilities. Interestingly, one project of Applequist's that he published in 1964 was the preparation of a putative precursor of a “Dewar benzene” system 15, see eq. 16. Pyrolysis of 30 produced “anthracene and bianthryl, but no indication of the anticipated dibenzobutalene”.170 Thus, Applequist was attempting to use electrocyclizations to produce what he termed “a nonbenzenoid aromatic or pseudoaromatic compound.”170 Also, Applequist had studied the photochemical dimerization of anthracene,170 a photochemical [4+4] cycloaddition pointed out by Woodward and Hoffmann in their first cycloaddition paper.171
Lastly, even though he had been a graduate student of Roberts, one of the experts in molecular orbital theory and its applications to organic chemistry in the 1950s and early 1960s, there is no evidence that Applequist had picked up on this technique. For example, there is almost no use of molecular orbital theory in his course on small ring chemistry at Harvard in the spring of 1964, according to Hoffmann's extensive notes of that course.163
3.11. Richard K. Hill (1928–)
3.11.1. Gateways
In 1964, Richard Hill (Figure 22) published two papers on what would later be termed sigmatropic rearrangements, one on the Claisen reaction172 and the other on the Alder-ene reaction173 (Figure 23). In both of these papers, Hill referred to “no-mechanism reactions.” Both of these papers are entitled, in part, Stereochemistry of ‘No-Mechanism’ Reactions. In these papers, Hill cited Sara Jane Rhoads's review/compilation paper136 on the no-mechanism reaction. In 1961, Hall had also published a major review on the stereochemistry of the prototypical cycloaddition reaction, the Diels-Alder reaction.174

Richard Hill, ca. 1965. Photograph courtesy R. Hill.

3.11.2. Why and Why Not Hall?
Why? Hill was primed to discover the Woodward-Hoffmann rules. He knew of the “no-mechanism” group of reactions. He had studied several of them, including cycloadditions and sigmatropic rearrangements in detail, especially their stereochemistry.
Why not? In his own words:
“The reason I never proposed orbital symmetry explanations is embarrassingly simple. I knew nothing about molecular orbital theory. MO theory wasn′t taught at Penn State in the 1940s or at Harvard in graduate school. Of course, other folks who were even older, and they made it a point to learn it on their own. But I have never had much mathematics ability beyond algebra, and in the 1960s, I was still primarily focused on natural products and synthesis. I became interested in cyclic transition states, suggested earlier by Charles Hurd, and I was fascinated with the idea of transferring chirality from one center to another. The paper with Mordecai Rabinowitz (who went on to become a distinguished professor at the Hebrew University of Jerusalem) was our first, and we went on to do similar studies in Claisen, Cope, and Stevens rearrangements. So to answer your question, my ‘blind spot’ was mathematical ignorance.”175
After reading an advanced draft of this paper, Hill shared the following memory.
“A year or so after the W−H rules were published, I was sitting in my Princeton office when the phone rang. A voice said, ‘Dick, this is Bob Woodward calling.’ I leapt from my chair, saluted, and stammered, ‘Sir! Yes, Sir! Professor Woodward, Sir!’ It felt like a phone call from God. My student T.-H. ‘Bill’ Chan had done a brilliant job demonstrating the transfer of asymmetry from nitrogen to carbon in the Stevens Rearrangement,176 and Bill was now at Harvard as a postdoc with Woodward and had presented a seminar on his work. RB thought our reaction might be subject to orbital symmetry and was calling for more details.”177
And there was another reason, shared by almost all the other chemists discussed in this publication. In the early 1960s, he was busy with other research projects, even though some – stereochemistry other possible pericyclic reactions, e. g., the Stevens rearrangement178 and the Beckman rearrangement179, 180 – were related to concerted cyclic processes.
3.12. Charles L. Perrin (1938–)
3.12.1. Gateways
Charles Perrin (Figure 24) received his undergraduate (1959) and Ph.D. (1963) degrees from Harvard. After postdoctoral studies at the University of California at Berkeley with Streitwieser, he joined the Department of Chemistry at the University of California at La Jolla.181 According to Perrin,

Charles Perrin, ca. 1967. Photograph courtesy C. Perrin.
“Frank Westheimer was my Ph.D. advisor. Actually, the person I really would have liked to work with as a graduate student was the physical/theoretical chemist William Moffitt, whose fabulous thermodynamics course I also took my senior year at Harvard, but who died that year at age 33. So I was also a physical chemist, and I took cumes in both organic and physical chemistry.”182, 183
According to SciFinder, Perrin's first publications were as follows: in 1966 on methoxy exchange of camphor dimethyl ketal;184 in 1968, on Applications to Spin-Spin coupling with n Equivalent Nuclei;185 in 1969 on the Claisen and Cope reactions186 and on vibronic coupling in polyenes and porphyrin;187 in 1970, a book entitled Mathematics for Chemists;188 in 1971, two papers on electrophilic aromatic substitution;189, 190 and in 1972, a pedagogical paper entitled The Woodward-Hoffmann Rules – An Elementary Approach.191
Perrin has had a distinguished academic career and in 2015 received the James Flack Norris Award in Physical Organic Chemistry. This slice of Perrin's biography is reported here to demonstrate that he is just one of many organic chemists – not just physical organic chemists – who were graduate students in the late 1950s or early 1960s. These young chemists, of whom due to Sputnik there were many, had not yet become so focused on their individual research programs to prevent them from being interested in the breadth of organic chemistry of that era. Any of them could have solved the pericyclic no-mechanism problem had they the prerequisites, as they all had the intellectual prowess to do so.
As Perrin recently told his story,
“One of the intellectual triumphs of organic chemistry is the Woodward-Hoffmann rules for pericyclic reactions… I regret that during the [famous Woodward] Thursday night seminars that I attended, and I left Harvard in February 1963, Woodward never disclosed his puzzlements regarding pericyclic reactions. I may have missed a seminar where he did disclose his puzzlements, or I may not have recognized the point of such a disclosure, but as far as I know, he did not. I had read Streitwieser's book on MO theory,192 so I would have recognized the relevance of aromatic transition states, as subsequently presented by Dewar103 and Zimmerman.101, 102”181, 193
As witnessed by Perrin's 1970 book Mathematics for Chemists188 and from his publications, Perrin was a physical-physical organic chemist. But according to his own telling, Perrin needed someone to describe the pericyclic no-mechanism problem to him. Perrin, as for many others, had the tools and the intelligence to solve the problem. He did not, however, know of the problem.
On several occasions after Perrin had left Harvard (in February 1963),194 Woodward discussed Vogel's mysterious reaction (eq. 5).39 And in March 1961, Woodward even included Vogel's reaction in a cumulative examination for early-term graduate students.195 So, some Harvard graduate students did know of the problem, or at least, they had been exposed to Vogel's reaction. One can only imagine one of the graduate students who took that March 1961 cumulative exam later deriving a MO explanation and publishing the solution on his or her own. But that did not happen.
3.12.2. Why and Why Not?
Why? Perrin is perhaps the best example of an extremely bright organic chemistry graduate student of the early 1960s with a very serious proclivity toward physical chemistry and theoretical chemistry. His postdoctoral studies were with Streitwieser at Berkeley, one of the premier theoretical organic chemists of that era and himself and expert in MO theory.192
Why not Perrin? Most likely, because he did not have knowledge of the pericyclic no-mechanism problem.
3.13. Jean-Marie Lehn (1939–)
3.13.1. Lehn: Ranganathan, Calculations for Woodward, and the No-Mechanism Puzzle
Graduate students and postdoctorals of the 1950s and early 1960s were not isolated from the events being discussed in this series. Indeed, several of them either were participants in developing the key leads (e. g., Subramania Ranganathan; see Paper 6 in this series4) or had front row seats on the podium, being so close to the events themselves.
Who were these graduate students or postdoctorals? Hoffmann was one. As also discussed in Paper 6 in this series, Hoffmann was a Harvard Junior Fellow – a special type of postdoctoral fellow – when Ranganathan was discovering his mysterious reactions. Ranganathan was Woodward's postdoctoral student who discovered the alternating stereospecific reactions while pursuing the oxazole route to vitamin B12 (Figure 25).105, 196 Jos Schlatmann was the co-author of the Havinga paper in which Oosterhoff's December 1960 suggestion was published.40, 197 Alfred Hortmann, as discussed in Paper 6 of this series,4 had lectured on Oosterhoff's suggestion to the Corey group. And there were others, many others. Indeed, for each faculty member discussed in Paper 6 of this series,4 many of their own graduate students and postdoctorals could have, even might have solved the pericyclic no-mechanism problem.

Another one of these was Jean-Marie Lehn (Figure 26) who 25 years later would receive his Nobel Prize in Chemistry. Lehn received his Ph.D. in France studying with the natural products chemist Guy Ourisson at the Universite de Strasbourg. Lehn's Ph.D. involved the NMR analysis of triterpenes and a spin-spin coupling problem. He arrived as a postdoctoral with Woodward in October 1963 and was “developing a camphor-derived component for the total synthesis of Vitamin B-12.”198 Hoffmann and Ranganathan were Lehn's first American friends.199

Jean-Marie Lehn examining NMR spectra, likely taken on a Varian A-60, Harvard University, early 1960s. Photograph courtesy Douglas Young.
3.13.2. Gateways
Several happenings involving Lehn occurred (or did not occur) during the first half of 1964 that are important to note.
Lehn did not sit in on Applequist's course on small ring compounds that contained many of the no-mechanism hints. But he did take John Baldeschwieler's course on quantum mechanics.200 This aligns with Lehn's interest in physics while in graduate school.
Lehn received help from Jack Gougoutas (the X-ray crystallographer), using the IBM 1620 in Harvard's computer center. Lehn did some computations with Gougoutas.198, 199 According to Lehn,
“I often talked with Subramania Ranganathan (very good friend) [whose laboratory was next door to Lehn's] about his results on thermal and photochemical reactions triene/cyclohexadiene interconversion in the TCK B-12 synthesis route.”200
Ranganathan certainly discussed his research with Lehn, and very possibly told him about the ring opening and ring closure during the synthetic work on vitamin B12, a different stereochemical course from that predicted by Woodward.199 That being said, Lehn returned to France in September or October 1964, perhaps before Ranganathan had completed his structure elucidations.
According to Douglas Young,
“I was at Harvard for two years (September 1963 to August 1965) and in the first of these years, shared a two-man lab with Subram Ranganathan. We knew him as Subram at Harvard but on trips to India, I noted that his Indian colleagues called him Ranga. Jean-Marie Lehn was working in the two-man lab next door to us and was friendly with Roald Hoffmann. My understanding is that when Woodward and Jean Marie discussed the possible theoretical basis for the reactions which eventually gave rise to the rules, it was Jean-Marie who introduced Hoffmann as being the person to carry the theoretical side forward.”201
According to Lehn,
“One evening RBW came in and jotted down on a yellow pad his ideas on the stereochemical features of thermal and photochemical reactions of dienes and trienes. [Woodward's graphic is shown in Figure 27.] There were not orbital symmetries involved. It was probably in Spring 1964. I do not remember the date.”199, 200, 202

Chemical pictography drawn by R. B. Woodward for J.-M. Lehn, spring of 1964. This displays several of the key reactions that were hints to the solution of the pericyclic no-mechanism problem. The JACS reference at the bottom is to the Corey-Hortmann synthesis of dihydrocostunolide,166 other examples of alternation of stereochemistry due to thermal/photochemical reactions. “In Lehn's drawing, the four lines [lower right-hand corner] make you think of butadiene MOs, but the discussion in remainder of page is all on hexatriene cyclization. Puzzling.”203 “The upper right hand corner,… HX additions.”204 The names Vogel39 and Fonken205 refer to researchers who studied the cyclobutene ring opening and the cyclohexadiene systems, respectively (eq. 5). At the very bottom, right corner is “1964 1st Semester”. Unfortunately, the date that this document was created was not inscribed. Graphic courtesy J.-M. Lehn.
On another occasion, Woodward asked Lehn “to get him the molecular orbitals of 1,3,5-hexatriene. I found them, gave them to Woodward.”199 According to Lehn,
“RBW asked me specifically to provide the hexatriene orbitals. I think that I did not compute them, but got them from the literature, but I am not 100 % sure.”198
and
“I mentioned to Roald what Woodward had asked me to do [the MOs of 1,3,5-hexatriene] and Woodward's writing on the yellow page and suggested that he speak with Woodward. Roald subsequently went to see Woodward and they talked about this chemistry.”199
Lehn did some extended Hückel calculations in 1964.206, 207 Lehn's laboratory partner Ian Fleming remembers Lehn carrying around large stacks of computer output, the typical output of computer calculations of the 1960s–1980s.208, 209 Almost certainly, Hoffmann gave the eHT program to Lehn210 and presumably helped him use it. Lehn recalls calculating either the thermal or the photochemical [2+2] cycloaddition of “two ethylenes to a cyclobutane with Hoffmann, not with or for Woodward.211 As recalled by Lehn,
“Despite the fact that I did not find any written notes [in 2015], I remember quite well that I calculated the face-to-face approach of two ethylene molecules.”212
According to Ranganathan,
“I am pretty sure of my statement that Jean-Marie did the very first calculations.”213
Lehn's memory is confirmed by Corey. In his November 2, 1981, letter to Hoffmann recounted, Corey wrote,
“In fact, [on May 4, 1964, Woodward] told me that some calculations just made for him by Jean-Marie Lehn showed that quantum mechanical orbital effects could not explain the experimental data. He went out to Jean-Marie's lab looking for him, but Jean-Marie was not around so this was not pursued further.”214
In 2011, Corey confirmed this in writing to Hoffmann, saying,
“Jean-Marie confirmed for me several years later that he had done some calculations for Woodward, but that the ‘calculations had blown up.’”215
On May 21, 1964, Woodward wrote a letter of recommendation for Lehn to Ourisson at the Universite de Strasbourg where Lehn would shortly take his first academic position and where he would remain. Woodward wrote in part,
“Lehn has also shown the breadth of his interests through seizing the opportunity here of delving into quantum mechanics.”216
Woodward added,
“The only respect in which the impression Lehn has made is not outstanding is in his manipulative ability in the laboratory, which still needs further development. I am sure there is no fundamental problem here, and that as time goes on he will develop a facility in that respect commensurate with his other qualities.”216
Lehn surely justified Woodward's anticipation. Lehn received the Nobel Prize in Chemistry in 1987.
3.13.3. Why and Why Not Lehn?
Why? We know that Woodward approached Lehn before Woodward approached Hoffmann, as Woodward was asking many of his students about the Vogel reaction (eq. 5). This was a topic that Woodward discussed at many of his Thursday evening seminars. We have Woodward's own words, that he asked Lehn to provide some quantum chemical information on the topic. We know that Lehn did some extended Hückel molecular orbital calculations, surely with some assistance from Hoffmann and certainly using Hoffmann's eHT software, and according to Lehn, not associated with Woodward's request for information about the molecular orbitals of 1,3,5-hexatriene. We know that Lehn had interacted with Ranganathan about his mysterious reactions (Figure 25).
Why not? By late 1963 and early 1964, Lehn was heavily occupied with his work on the total synthesis of vitamin B12 and also anticipating and preparing for his return to Strasbourg and the beginning of his own independent academic career. His mind was not on identifying new major research programs outside his then vision. There was one other likely reason why Lehn did not discover orbital symmetry control. As Lehn recently said, he “does not jump on the research problems of others.”217
3.13.4. After Harvard
Lehn's interest in pericyclic reactions continued after his departure from Harvard and into his independent career in Strasbourg that began in the summer of 1964. In a May 1965 letter to Hoffmann, Lehn described some of his research interests (Figure 28). Lehn was fascinated by what was later termed periselectivity by Houk, i. e., the bifurcation among two or more Woodward-Hoffmann allowed reactions.218, 219 This contrasts with the key alternation hints used to solve the no-mechanism problem.

Excerpt from a letter by Jean-Marie Lehn to Roald Hoffmann dated May 22, 1965. Lehn, in his first year of his academic appointment at the Universite de Strasbourg, described some of his research plans. Lehn wrote, “Furthermore, I am interested in the effect of conformation and stability of the excited states on the course of photochemical reactions…[220]” This was a topic of interest to several others who were close to solving the no-mechanism problem, such at Egbert Havinga at Leiden and William G. Dauben at Berkeley. Both Havinga221, 222 and Dauben223, 224 studied what they described as ground state control of photochemical reactions. See Paper 3197 and Paper 64 in this series for more details on Havinga and Dauben, respectively.
3.14. Jerome A. Berson (1924–2017)225-227
3.14.1. General Background Information
Jerome Berson (Figure 29) received his B.S. degree from the City College of New York City in 1943. After six months as an assistant chemist at Hoffmann-LaRoche in Nutley, NJ, he served two years in the U.S. Army. He completed his Ph.D. at Columbia University with Doering in the Spring of 1949. After a National Research Council fellowship with Woodward, he joined the University of Southern California as an Assistant Professor in September 1950. Berson was a Guggenheim Fellow at Caltech in the Spring and Summer of 1959. In 1963, he moved to the University of Wisconsin – Madison, and six years later, in 1969, moved to Yale. Berson was a leading physical organic chemist.

Jerome Berson, ca. January 1950. This photograph was sent to the University of Southern California with his job application. Photograph courtesy J. Berson.
3.14.2. Gateway: Pericyclic Reactions
In 1967, Berson and Gordon L. Nelson published what may be the most demanding test ever of the Woodward-Hoffmann rules, namely Inversion of Configuration in the Migrating Group of a Thermal 1,3-Sigmatropic Rearrangement.46, 228 But years before his probing of the validity of these rules, Berson could well have developed them himself.
As a graduate student with Doering, Berson was exposed to aromaticity and likely to Doering's developing ideas of aromaticity and the 4n+2 rule, first proposed as such by Doering and Berson's fellow graduate student Francis Detert in 1951.92 Two years before the 4n+2 rule, on May 7, 1948, Berson gave a Columbia organic symposium on The Tropolones,229 At that time, tropolones were the first class of nonbenzenoid compounds identified as aromatic. The typewritten structures of stipitatic acid230 and purpurogallin231 (Scheme 2) are excerpted from Berson's lecture. Note the carelessness in the chemical pictography which is atypical of Berson; but he was a young student. The same observation – carelessly drawn chemical structures from an individual who later became known as a perfectionist in drawing structures232 – can be seen from R. B. Woodward's undergraduate thesis233 and especially his Ph.D. dissertation.234

Structures of stipitatic acid and purpurogalin as drawn by and presented in Berson's 1948 Organic Chemistry Seminar handout.229 At that time, these two structures and that of tropolone were proposed by Dewar230 and Holger Erdtman231 but the resonance stabilization of a six-electron π-system shown awaited Doering's 1951 paper.92
In the first 15 years of his independent academic career, Berson published 74 papers. Beyond various projects involving natural products chemistry, Berson's primary focus was physical organic chemistry, especially the mechanism of reactions. And Berson's knowledge of MO theory was immense, especially when compared with his peer group. As Hoffmann commented after reviewing Figure 30,

Excerpts from Jerome Berson's 1960 notebook while at the University of Southern California.235 Of note is Berson's use of the Hückel molecular orbitals of butadiene coupled with Kenichi Fukui's frontier molecular orbital method (see top line including Fukui's seminal 1952 FMO paper where Berson writes “frontier orbital” and “frontier electrons”) to predict the regiochemistry of attack by a carbocation.
“That's an amazing notebook page from Berson in 1960. It's all there! How could someone know all this – the MOs and the idea of frontier orbitals, and not go on to solve the problem. I'm not faulting Berson; I liked him so much as a person and scientist. I think Berson understood MO theory.”86
In the 1950s and early 1960s, many of Berson's publications dealt with what later would be called pericyclic reactions. Berson's major thrust was the Diels-Alder reaction resulting in at least 12 publications between 1953 and 1965 on that cycloaddition. Some of the titles of those papers are Stereochemistry and Mechanism of the Diels-Alder Reaction (1956)236 and The Mechanism of the Diels-Alder Reaction (1961)237 and On the Mechanism of the Indene-Maleic Anhydride Reaction (1964).238 Also in that time period, Berson reported on various sigmatropic rearrangements,239-242 several of which are shown in Figure 31.

3.14.3. Assembling the Experiences
-
Berson was quite familiar with MO theory. In a set of “MO notes” dated by Berson for this author as having been written ca. 1960,243 Berson outlined the fundamentals of LCAO theory, calculated using matrices the HMOs of 1,3-butadiene, derived the MO energy levels of a series of conjugated monocyclic systems, and then used frontier MO theory, citing Fukui's seminal 1952 paper,244 and finally used group theory and symmetry properties to derive a state diagram (not shown in Figure 30).235 In 1964, in a paper entitled On the Criteria for Pi-Electron Delocalization in Some Cyclic Conjugated Systems,245 Berson et al. reported Hückel molecular orbital calculations and modified HMO theory (ω-technique) and calculated charge distributions and dipole moments. This work was designed to evaluate the use of molecular dipole moments and 1H NMR to estimate the extent of π-electron delocalization as criteria for aromaticity. And prior to 1964, Berson spent part of a semester on sabbatical at Caltech. “One of my objectives was to attend some of Jack Roberts's lectures on MO theory. Also Streitwieser's book came out about then, so I was trying to organize everything into a manageable package for myself and the students, probably starting at USC.”243
Was Berson aware of the no-mechanism problem? There are several pieces of evidence that indicates a strong “yes.”
-
In the winter of 1959, Berson attended a Woodward Thursday evening seminar, during which Woodward presented a discussion of Vogel's mysterious reaction (eq. 5).1, 243
-
In a March 6, 1962, letter to Berson,246 Melvin Goldstein (Cornell) presented a short discussion on the two valence isomerizations shown in Figure 32
 Figure 32
Figure 32Graphical excerpt from Melvin Goldstein's March 6, 1962, letter to Jerome Berson.246
. Here again we see the famous Vogel reaction (eq. 5).
-
Berson was the discussion leader for Doering's lecture at the June 1964 Reaction Mechanisms Conference on Valence Tautomerism and No-Mechanism Reactions where the Vogel and Criegee reaction was discussed as an anomaly.81
-
Doering gave a seminar at the University of Wisconsin in the spring of 1964 discussing the no-mechanism problem. Immediately after Doering's lecture, Berson spoke with Doering, his former Ph.D. advisor, and suggested an aromaticity-antiaromaticity explanation. In Berson's words,

“Doering gave a lecture on cyclobutene ring-openings, making proper reference to both Vogel and to subsequent work by Criegee (eq. 5). As I recall, he made some statement, based just on crude visual examination of molecular models, that he thought disrotation should be preferred. He was quite puzzled that conrotation was the result instead, and he left this question unresolved. At the end of the lecture, I went up to him and asked him whether he thought that the transition state for the disrotatory path might suffer from the same thing that was wrong with cyclobutadiene, namely antiaromaticity. He seemed interested in the idea but we did not pursue the point. On the way out of the lecture room, Howie Zimmerman, who had not been a party to our conversation, button-holed Bill and asked him the same question, and Bill told him that I had raised the same point.”247
Thus, in the spring of 1964, Berson (and Zimmerman) related the disrotatory transition state of an electrocyclization to cyclobutadiene destabilization. There is no need to debate when the concept of “antiaromaticity” (and that actual term) first appeared in the chemical community. Rather, the destabilization of CBD was known well before 1964, if not in the early 1930s by Hückel.248-251 It is an important example of a forerunner, a good piece of the truth, one that could have been parlayed by Berson along with his knowledge of MO theory and many experimental excursions in valence isomerizations into his developing the orbital symmetry story before Woodward and Hoffmann.
At what stage did Berson get a deep understanding of the Woodward-Hoffmann rules? As Berson related,
“We had also been working on the mechanism of the Diels-Alder reaction, specifically on whether it was concerted. That led us to set up an experiment to test whether the energy surfaces of the Diels-Alder and the 1,3-sigmatropic rearrangement are connected. That led us to the series of experiments to test orbital symmetry culminating in George Nelson's demonstration that the reaction went with inversion of the migrating carbon.46 The design of that experiment took place in about 1964–1965 and forced us to think about orbital phases. So that‘s how we finally got there.”243
3.14.4. Why and Why Not Berson?
Why Berson? Berson was one of the most intellectual and clever physical organic chemists of the era. Ultimately, in 1967, it was Berson who provided perhaps the strongest and most demanding chemical example supporting the orbital symmetry concept.46 His analyses of the mechanism of the Diels-Alder reaction is classic in their design and interpretations.237 He had been studying the concertedness-nonconcertedness bifurcation of the Cope reaction in the early 1960s.240 He was a scholar of valence isomerizations.
Why not Berson? Here are some reasons.
-
Berson and his group moved from the University of Southern California in 1963, thereby creating a distraction in the flow of Berson's research program.
-
Berson was very much focused on specific slices of chemistry in the early 1960s, e. g., the mechanism of the Diels-Alder reaction and the mechanism of tandem valence isomerizations, not on looking for generalizations.
-
In Berson's own words:
“If your question is, ‘Why did it take us so long’? I′d have to give you the same answer I once received from Friedrich Hund when I asked him why he missed out on the physical explanation for Hund's Rule. He replied that the explanation should have been in his first paper in the field, ‘Aber so klug war ich eben nicht.’ [‘I was just not that smart.’]”243
On another occasion, Berson responded to the same question,
“So we have to live within our capacities… Woodward was simply smarter than any of the rest of us.”243
And why did Berson not jump right in and solve the cycloaddition problem or the sigmatropic rearrangement problem immediately after the first Woodward-Hoffmann paper on electrocyclizations was published in January 19655? As Berson explained,
“The answer to your question about ‘jumping right in’ is that I was an experimentalist. Experiments take time. So, for example, the first results of George Nelson's work on sigmatropic rearrangements did not come out until 1967. Even that work depended on some earlier work in our lab by Bob Wood and Jim Patton, which was done about 1962–1965. So yes, we did ‘jump right in.’ Fortunately,68, 69, 252 we had prepared the ground for Nelson's findings.”253
3.15. John D. Roberts (1918–2016)134, 135, 254-256
3.15.1. General Background Information
By 1965 when the Woodward-Hoffmann rules were first published, John D. Roberts (Figure 33) had published well over 200 papers. He was one of the premier physical organic chemists in the second half of the 20th century. He began his independent career at MIT, having been invited there by Arthur C. Cope. Roberts then moved to Caltech at the invitation of Linus Pauling. He was also a noted textbook author including his best seller Roberts and Caserio (Basic Principles of Organic Chemistry257) and textbooks involving MO theory and nuclear magnetic resonance.129, 258, 259 What follows now are highlights from Roberts's pre-1965 research that relate to his predisposition to discover the W−H rules.

John D. Roberts at MIT, 1947. Photograph courtesy J. D. Roberts.
3.15.2. Gateway: Pericyclic reactions
In 1951, Roberts et al. reported that the solvolysis of cyclopropyl p-toluenesulfonate produces the rearranged product allyl acetate (eq. 17). They wrote,
“While it is not certain that the cyclopropyl cation is actually an intermediate in these reactions, the very low reactivity of cyclopropyl p-toluenesulfonate makes it unlikely that the relatively stable allyl cation is formed directly in the rate-determining step.”260
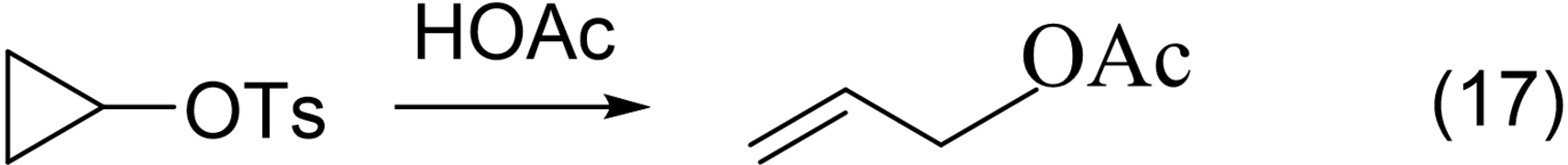
This reaction is the prototypical two-electron electrocyclization discussed by Woodward and Hoffmann in their first paper.5 See Paper 6 in this series for a discussion of the research of Charles H. DePuy, Stanley J. Cristol, Christopher S. Foote, Wolfgang Kirmse, William E. Parham, Roland Pettit, Paul von R. Schleyer, and Philip S. Skell who examined the relative reactivites and stereochemical consequences of solvolyses of various cyclopropyl-X derivatives.4
In the early 1960s, Roberts published many papers on the mechanism of the Diels-Alder reaction.261-264 These papers primarily studied the mechanistic details, e. g., electronic factors, of the Diels-Alder and retro-Diels-Alder reaction. Citing Woodward and Katz's 1959 research on Diels-Alder dimers, i. e., “products of Diels-Alder additions in which a single component serves both as diene and dienophile,”263 Roberts and Raymond P. Lutz determined the rate constants of partial dissociation  re-association and complete dissociation
re-association and complete dissociation  Diels-Alder reaction of the optically active 16 to possibly (slightly) racemized 16 and 17 (eq. 18) They concluded,
Diels-Alder reaction of the optically active 16 to possibly (slightly) racemized 16 and 17 (eq. 18) They concluded,
“While our experiments offer no new insight into the mechanism of the Diels-Alder reaction, they exclude participation of a diradical intermediate with a long enough lifetime to permit racemization. Strong moral support is provided for the presumption that the rearrangements and the reverse Diels-Alder reaction proceed along the same energy path. The possibility that different energy paths are involved (the rearrangements are formally Cope rearrangements) is not rigorously excluded; but at least with methacrolein dimer, both reactions occur simultaneously.”263

This conclusion is interesting in that the title of Roberts and Lutz’ paper is Mechanism of the Diels-Alder Reaction.263
In 1965, Roberts, Camille Ganter, and Ulrich Scheidegger determined that reacting 18 in t-pentylbenzene at 190 °C results in only external rearrangement to 19 rather than internal rearrangement.262 “External” refers to a retrogression of the endo adduct to maleic anhydride and cyclopentadiene (eq. 19). “Internal” refers to a mechanism not involving complete dissociation into kinetically free maleic anhydride and cyclopentadiene. Berson had previously found both internal and external rearrangements when the reactions were performed in decalin also at 190 °C.236, 265
Also in 1965, Roberts and Joseph Lambert assessed the possibility of a two-step Diels-Alder reaction by using a diene that was “heavily weighted toward a diradical pathway.”261 To compliment this strategy, they synthesized a subtly-substituted dienophile designed to reveal a loss of chirality should a diradical pathway obtain (eq. 20). Complete stereospecificity was observed.
In the mid-1950s, according to his autobiography, Roberts convinced Pauling, then Chair of Caltech's Department of Chemistry, to provide funds to purchase Caltech's first NMR spectrometer,
“then operating at 30 MHz … As my punch line, I told him that ‘with NMR, we can investigate the borderline between resonance and tautomerism.’ It seemed to spark his interest, and I explained how we could study cycloheptatriene as a function of temperature (Scheme 3)… ”135

Two of the key hints that led to the Woodward-Hoffmann rules were Emanuel Vogel's39, 267 and Rudolf Criegee's16 stereospecific ring opening reaction of cis-3,4-disubstituted cyclobutenes to (E,Z)-1,3-butadienes (eq. 5). Roberts published a number of papers on the physical and chemical properties of cyclobutenes,268-270 many of which were aimed at preparing derivatives of cyclobutadiene (CBD). This is reported in the following subsection. Thus, Roberts was an expert in cyclobutene chemistry.
Putting all this together, Roberts was an expert in many types of valence isomerizations and cycloadditions. He was also an expert in small ring chemistry, from which most of the orbital symmetry hints derived.
3.15.3. Gateway: Aromaticity
In molecular orbital calculations reported in 1952271 and 1958,272 Roberts pointed out that the lowest (ground) state of cyclobutadiene was predicted to be a triplet.
In Roberts, Andrew Streitwieser, Jr., and Clare M. Regan's 1952 paper, they came tantalizingly close to the concept of antiaromaticity,130 a concept that was not unambiguously pronounced until 1965.98-100 Their MO calculations revealed a stability of 4n+2 monocyclic hydrocarbons and an instability of 4n systems. See Scheme 4. They wrote,

An excerpt from Roberts, Streitwieser et al.’s 1952 paper illustrating the stability (DE=delocalization energy).130 The DE for cyclobutadiene was calculated to be zero. Reprinted (and adapted) with permission from J. D. Roberts, A. Streitwieser, Jr., C. M. Regan, J. Am. Chem. Soc. 1952, 74, 4579-4582.. Copyright 1952 American Chemical Society.
“The apparent instability of [cyclobutadiene] might be ascribed to the triplet ground state on the basis of the molecular orbital treatment since the known cyclopropane should have comparable or greater angle strain.”130 [emphasis added]
Roberts published several papers aimed at synthesizing derivatives of cyclobutadiene, testing the limits and characteristics of aromaticity. Heating the optically active cyclobutenone 20 led not to the cyclobutadiene derivatives 21 but rather to ring opening, as demonstrated by the capture of the ketene with ethanol (eq. 21).268, 270, 273 That ring opening is a retro-electrocyclization.
In 1955, Roberts and Edgar J. Smutny reported the synthesis of phenylcyclobutendione.274 They wrote,
“The failure to detect cyclobutadiene or its non-fused ring derivatives among the products of appropriate synthetic reactions can be ascribed either to excessive ring strain or unfavorable electronic configurations. Strong evidence that ring strain is not the most important factor is now provided by a synthesis of the quite stable phenylcyclobutadienoquinone which to a first approximation is expected to have the same degree of ring strain as phenylcyclobutadiene.”274
By 1955, there was sufficient theoretical considerations that cyclobutadiene was not aromatic, indeed was destabilized relative to 1,3-butadiene, for reasons best understood using quantum chemical theory.251, 275-278 Recall also that it was in 1951 that William von E. Doering invoked the “2+4n” rule92 following up on Hückel's MO explanation of the 1930s.248, 250, 251, 279
Roberts's research was cited by Criegee in reference to literature precursors to cyclobutadiene.280 This was a high compliment, as it was Criegee who first made CBD-metal clusters.281
In 2009, Roberts reminisced,
“For many years, we had ambitions of preparing cyclobutadiene or derivatives thereof.”134
Aligned with the instability of cyclobutadiene, Roberts published extensively in [4+2] cycloadditions – Diels-Alder reactions – and had often demonstrated that they were thermally facile and fit within the no-mechanism reaction scheme. He was also an expert in [2+2] cycloadditions;282, 283 in 1962, Roberts and Clay M. Sharts published a chapter in Organic Reactions entitled “Cyclobutane Derivatives from Thermal Cycloaddition Reactions.284 Roberts knew that these were radical reactions, that a four-electron transition state was unfavorable while a six-electron transition state was favorable.
Roberts's book on molecular orbital calculations (see below)129 has a chapter on aromaticity.
3.15.4. Gateway: Molecular Orbital Theory
In 1952, Roberts and Streitwieser used Erich Hückel's molecular orbital method to calculate the orientation patterns in electrophilic, radical, and nucleophilic aromatic substitution reactions.271 In that same year, Roberts, Streitwieser and Regan used the LCAO (linear combination of atomic orbitals) method to calculate the properties of small-ring hydrocarbons (discussed in the section immediately above).130
In 1955, Roberts and Dorothy A. Semenow calculated the charges on the amino nitrogen of 4-substituted anilines using the Hückel molecular orbital method.285
In 1956, Roberts, William G. Woods and Rudolph A. Carboni used an LCAO method to calculate the delocalization caused by the olefin moiety in the solvolysis of anti-7-chloronorbornene as reproduced from their paper in Scheme 5.132 In order to treat this non-planar system using the Hückel method, the π orbitals were bonded in a σ fashion, i. e., for the calculations, they were geometrically and algebraically modified such as in Figure 34(A)). In other words, Roberts was calculating molecular orbitals with basis sets containing only π-orbitals, not the entire molecule (i. e., not σ-orbitals). Referring to his 1952 paper with Streitwieser, Roberts pointed out the relationship of the “homoallylic” or “bis-homocyclopropenyl” carbocation to cylopropenyl carbocation, noting that these are both 4n+2=2 electron stabilized systems (where n=0) (Scheme 5).

Graphical excerpt from Roberts, et al.132 showing π-delocalization during the solvolysis of anti-7-chloronorbornene.

Graphical excerpts from John D. Roberts's 1961 book on molecular orbital theory.129 (Top) A geometric model used to perform LCAO calculations for non-planar systems. This approach allows to the calculation of nonplanar systems. Thus, it is related to the extended Hückel method developed in the Lipscomb group by Roald Hoffmann and others286, 287 and parallels a method published earlier by Massimo Simonetta and Saul Winstein in 1954.288 (Bottom) Molecular orbitals for 1,3-butadiene from Roberts's above cited book.
In 1963, Merlin E. H. Howden and Roberts133 used LCAO calculations following a method used previously by Simonetta and Winstein288 for non-planar systems by using p-orbitals bonded in a σ fashion (Figure 35; see also Figure 34(Top)).

Graphic excerpt from Howden and Roberts's 1963 paper for one configuration of bicyclobutonium.133
Perhaps the single most important evidence of Roberts's expertise in theoretical and computational chemistry was his single-authored book Notes on Molecular Calculations published in 1961.129 Of relevance to the discovery of the Conservation of Orbital Symmetry, in that volume, Roberts performed the full Hückel calculations to obtain the molecular orbitals of 1,3-butadiene (Figure 34(Bottom) and also demonstrated a simple method to do MO calculations for non-planar systems (Figure 34 (Top)). Furthermore, Roberts discussed, though briefly, perturbation theory and Kenichi Fukui's frontier molecular orbital theory. As an example of the latter, Roberts wrote,
“For butadiene, the lowest unoccupied orbital has the wave function ψ3=0.6015χ1−0.3717χ2−0.3717χ3+0.6015χ4. The frontier orbital approach predicts, therefore, that nucleophilic attack on butadiene should occur at the 1 and 4 positions.”129
Hoffmann noted,
“Jack had to have courage to do MO theory IN THE HOUSE OF PAULING [who, of course, was a valence bond advocate], so to speak. Pauling was on his way out of mainstream chemistry by the time that MO work was done.”86
Thus, Roberts had the quantum mechanical knowledge of Hückel, LCAO and FMO theory, perturbation methods, and simple MO computational experience including calculations of non-planar systems. Without any doubt, Roberts had the theoretical chemistry and quantum chemistry knowledge to have published the first Woodward-Hoffmann paper in 1961, four years earlier than the Harvard team.
3.15.5. Why and Why Not Roberts?
Why Roberts? Roberts was one of the most well positioned organic chemists to solve the pericyclic no-mechanism problem. He was a small ring chemist. He was an expert in qualitative and quantitative MO theory. That is, he had performed MO calculations. Indeed, he had applied MO theory to chemical reactivity.
Why not Roberts? As Roberts wrote to Seeman in 2010,
“I was not working in the field, and while I knew in a general way about the symmetry of orbitals, it was not something I was thinking about when I wrote MO Calculations.”289
A year later, I questioned Roberts further. He responded,
“Pure SLOTH in keeping up with the literature and being busy with other things. Later at Caltech, I wrote with Clay Sharts a review on alkenes to cyclobutanes,284 I still had the blinders on. But more important and easier, why did not I invent MRI?”290
I then asked, “Perhaps you have not seen the photochemical results in the Vitamin D system? Was it simply that your attention was on other topics such as benzyne, MO theory, and NMR and writing your textbooks?” Roberts replied,
“YES!! And stupid narrowness in the literature I was reading.”290
Is it as simple as not reading the literature beyond one's own current research interests? Perhaps, but there is another data point: Roberts attended the 1964 Reaction Mechanisms Conference during which Doering lectured on the no-mechanism problem and presented his domino-motion explanation for the Vogel/Criegee cyclobutene ring opening (eq. 5).56 Indeed, Roberts presented the first lecture of that meeting on Magnetic Spectroscopy and Fast Reactions. Also, Sara Jane Rhoads, who had just written her major essay on the no-mechanism problem, spent a short sabbatical with Roberts in 1963.124 Roberts was also studying several electrocyclizations and was deeply involved in determining the mechanism of the Diels-Alder reaction. Roberts had studied aromaticity and anti-aromaticity. He had calculated the molecular orbitals of 1,3-butadiene and of cyclobutadiene.
But he was, indeed, a busy academic. In 1965 alone, he published 21 papers, just not on orbital symmetry. He was chair of the Department of Chemistry at Caltech in the early 1960s. He was also busy writing the breakthrough textbook Basic Principles of Organic Chemistry with Marjorie Caserio.257 One of Roberts's students described Roberts's competing stimuli as,
“During those years, we rarely saw him. There was one six-week period when I desperately tried to find him to discuss a research crisis, without success. Once a month we turned in our reports. He faithfully read them, covered them with red ink, and returned them, without any direct contact. That was our means of communication. We developed our own research projects, executed them, wrote the papers (more red ink), and when the referee reports arrived, they were redirected to us, who prepared responses and rebuttals. We remember that JDR hardly noticed the Cuban Missile Crisis. He certainly was not thinking about pericyclic reactions.”291
3.16. Andrew Streitwieser, Jr. (1927–2022)292-295
3.16.1. General Background Information
Andrew Streitwieser (Figure 36) was also in the right place at the right time to solve the pericyclic no-mechanism problem. The first decade of his career was as an experimental physical organic chemist. The middle decades melded wet chemistry with computational chemistry. The last decades, especially after his official retirement, he transformed into entirely computational chemistry.

Andrew Streitwieser, Jr., 1950s. Photograph courtesy A. Streitwieser, Jr.
Streitwieser learned about aromaticity and pseudoaromaticity and non-aromaticity by osmosis in graduate school (1948–1951). As documented in the section focused on Doering in Paper 6 of this series,4 Streitwieser recalled Doering discussing with him in 1949 “why ethylene does not dimerize to cyclobutane whereas it does react with butadiene to give cyclohexene.”296 It was also during Streitwieser's time with Doering that Doering invoked with Francis L. Detert the “2+4n” rule.92 Streitwieser's thesis research involved the methanolysis of the optically active phthalate derivative 22 in the presence of various solvent additives (eq. 22).297 The objective was to assess the stereochemical integrity of a displacement reaction about a tertiary carbon atom.298 This solvolysis reaction was to form the basis for many years of Streitwieser's independent research.299, 300
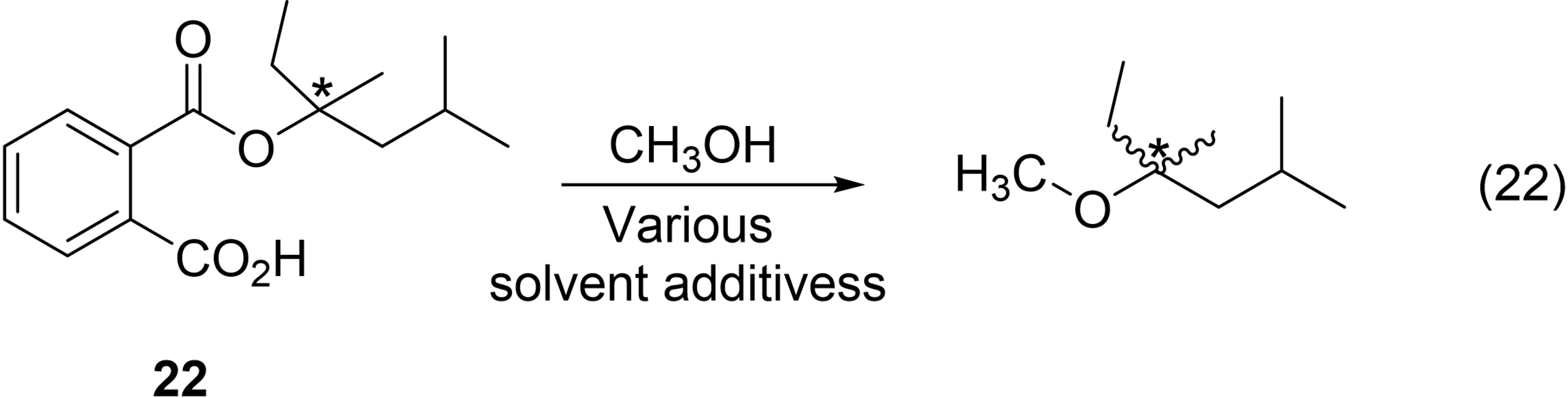
3.16.2. Gateway: Pericyclic Reactions
While he was knowledgeable about valence isomerizations and cycloadditions in the early 1960s, and there are many examples in his 1961 textbook,301 Streitwieser did no research in that area. It is likely that not being involved professionally in no-mechanism reactions was a serious limitation in his possibly solving the problem. On the other hand, Streitwieser was a close colleague with his Berkeley peer William G. Dauben who had enormous experience in pericyclic reactions, as discussed in Paper 6 of this series.4 As Streitwieser recalled,
“Bill Dauben did talk to me several times about the photochemistry of vitamin D [eq. 1 and eq. 2 in Figure 1] but I cannot recall being at all helpful to him in terms of theory and specifically in terms of orbital symmetry.”296
Almost certainly, had Dauben gone to Streitwieser just as Havinga went to Oosterhoff in December 1960,40, 197 we might have today the Streitwieser-Dauben (or the Dauben- Streitwieser) rules. How very strange that non-eponym sounds today.
3.16.3. Gateway: Molecular Orbital Theory
Streitwieser spent 1951–1952 as a postdoctoral student with J. D. Roberts at MIT. This was the period that Roberts was beginning his own interests in MO theory, and Streitwieser was his premier student at that time. Together with Roberts, Streitwieser published the paper discussed in the Roberts's Section 3.15 on MO calculations of small ring hydrocarbons. In 1952, Roberts and Streitwieser came very close to discerning antiaromaticity by very clearly distinguishing between 4n versus 4n+2 monocyclic hydrocarbons.130 Also in 1952, Roberts and Streitwieser used MO theory to calculate orientation in aromatic substitution reactions.271
Streitwieser also published several papers from MIT as the sole author. One was a molecular orbital computation of the ionization equilibria of triarylchloromethanes (eq. 23).302 In order to perform the calculations on these non-planar model compounds, Streitwieser performed simple LCAO molecular orbital calculations on each planar aromatic subunit, then added up the results assuming no interaction between rings. While this is a rather simplistic model, it demonstrates the clear need to perform MO calculations on non-planar substances and the approximations one could make as early as 1952 in order to do so.
In his first years at Berkeley, between 1952 and 1959, Streitwieser published 30 more papers, but only in the latter years did he return to publishing molecular orbital computations. All the other papers were wet chemistry, mostly dealing with solvolysis reactions (over 20 papers) and a series that he entitled Stereochemistry of the Primary Carbon (Part XIII appeared in 1964303), most of which involved solvolyses or closely related reactions (in addition to the 20 above mentioned papers).
In 1956, Streitwieser published a major review article on Solvolytic Displacement Reactions at Saturated Carbon.299 This was followed up in 1962 with a book entitled Solvolytic Displacement Reactions.300 In the late 1950s, he began to publish another series entitled Acidity of Hydrocarbons (Part XV was published in 1964303). Thus, for an academic who is mostly thought of today (and for the past several decades) as a computational/theoretical chemist, Streitwieser was primarily an experimentalist in his first two decades of his career.
In the early 1960s, Streitwieser returned to computational chemistry and molecular orbital theory. In 1960, he published An LCAO Treatment of Acidity of Hydrocarbons,304 a computational companion to his experimental papers on this topic. In 1962, with Irving Schwager, he used MO theory to calculate the polarographic reduction of aromatic hydrocarbons.305 Also in 1960 and 1962, Streitwieser used HMO to study ionization potentials.306, 307 The abstract of the 1962 paper is particularly interesting because it is an early example of trying to incorporate sigma bonds into a π-system calculation.
“The change in potential caused by introducing a methyl substituent into an organic compound can be represented by means of a simple model of a methyl group isolated from the π-system. Its electron-donating effect is recognized by making the attached π-C atom more electropositive.”307
With John Brauman, in 1963 Streitwieser developed a modification of Hückel molecular orbital theory to derive a novel index for nitration of aromatic compounds.308 Also in 1963 and again with Brauman, he used HMO theory to model the acidity of very weak acids.309
Perhaps Streitwieser's most important MO contribution in the 1960s was the publication of his 1961 book Molecular Orbital Theory for Organic Chemists.192 As R. Stephen Berry wrote in his book review that appeared in the Journal of the American Chemical Society,
“It seems, at last, that organic chemistry, even the whole of chemistry, has entered that golden age when chemists in all fields can begin to make effective use of quantum mechanics. . . the past dozen years have seen p-electron theory, once the exclusive plaything of physicists and physical chemists, used as a guide for essentially every field of organic chemistry. . . Professor Streitwieser has written a book to fulfill exactly this function – to give to the organic chemist the operating tools and a full scale of results, so that he can use the methods and have considerable feeling for the range of their validity.”310
In his book, Streitwieser discussed the MOs of 1,3-butadiene as an example. He set up its secular determinant, produced the third order determinants, obtained the roots for the quadratic equation, and calculated the coefficients for each of the four molecular orbitals. Streitwieser also drew the free electron wave functions as well as the Hückel molecular orbitals for 1,3-butadiene (Figure 37). From this diagram and either Vogel's39, 267 or Criegee's16 cyclobutene ring opening, Streitwieser could have written the first Woodward-Hoffmann paper.

The Hückel molecular orbitals for 1,3-butadiene from Streitwieser's 1961 book.192
Perhaps the most notable characteristic of Streitwieser's book is its discussion of chemical reactions. The book is divided into three sections: Part I. Simple Molecular Orbital Theory. Part II. Properties of Molecules. Part III. Reactions. While MO books of the 1960s and earlier focused almost exclusively on the theory and properties of molecules, Streitwieser's third section was over 150 pages in length and covered aromatic substitution, carbonium ions, radicals, carbanions, and of particular importance to Woodward-Hoffmann, it covered cycloadditions and rearrangements – though oddly in a section entitled Four-Center Reactions. While he discussed the Cope and Claisen rearrangements, Streitwieser did not cover any other valence isomerizations such as the 1,3,5-cycloheptatriene  norcaradiene or cyclooctatetraene
norcaradiene or cyclooctatetraene  bicyclo[4.2.0]octa–2,4,7-triene or the 1,3-butadiene
bicyclo[4.2.0]octa–2,4,7-triene or the 1,3-butadiene  cyclobutene rearrangements.
cyclobutene rearrangements.
A number of topics in Streitwieser's text are particularly relevant to the pericyclic no-mechanism problem.
-
Chapter 10 is entitled Aromaticity and the 4n+2 Rule. Streitwieser included Arthur A. Frost and Boris Muslin's graphic of Hückel molecular orbitals for monocyclic systems of size n=5, 6 and 7 illustrated as the vertices of k-fold regular polygons inscribed in circles with one vertex pointed downward (for n=3, see Scheme 6
 Scheme 6
Scheme 6(Left) Frost and Muslin's graphic for the energies (in β units) of the Hückel molecular orbitals (HMOs) for cyclopropane and its various charged analogues.311 (Right) Zimmerman graphic for the energies (in β units) of the Möbius configuration of HMOs for cyclopropane and its various charged analogues.101
, left).

Streitwieser used this mnemonic to illustrate the simple determination of the Hückel molecular orbital energy levels for monocyclic hydrocarbons. This mnemonic was used to demonstrate the particular stability of 4n +2 electron systems. Streitwieser spoke of “the instability of cyclooctatetraene”301 (not a 4n +2 electron system) and also wrote about the instability of cyclobutadiene:
“The cyclobutadiene ring structure has eluded a number of ingenious attempts at synthesis; these clever experiments provide definitive evidence of but little resonance energy in this system… [Several molecular orbital] models have no resonance stabilization, and this strained ring system should theoretically be an exceedingly unstable substance.”301
But Streitwieser did not classify 4n monocyclic systems as antiaromatic. Nor did he illustrate the Möbius (antiaromatic) system (Scheme 6, right) that Howard E. Zimmerman would use just a few years later to explain forbidden reactions using the orbital symmetry concept in pericyclic reactions.101
-
In Chapter 10, Streitwieser wrote,
“The cyclopropenyl cation, with two electrons, obeys the 4n+2 rule and should be stable. The cyclopropenyl radical and anion both have electrons in antibonding MO's; they also have unfilled shells and should be relatively unstable in theory.”301
But Streitwieser did not label 4n+2 compounds as aromatic; and he did not label 4n compounds as being antiaromatic. Indeed, in Chapter 10, he wrote
“that neutral hydrocarbons will be thermodynamically stable only if they have no electrons in antibonding MOs and have all bonding MOs completely occupied.”192
Streitwieser easily could have added, “and the converse.”
-
Chapter 15 discusses the [4+2] cyclization/Diels-Alder reaction and the [2+2] cycloaddition of two ethylenes (Figure 38
 Figure 38
Figure 38Excerpts from Streitwieser's book on Molecular Orbitals.301 “(Left and Center) Possible transition states for Diene Synthesis. (a) Geometrically symmetrical. (b) Geometrically unsymmetrical.”301 (Right) “Possible transition state for cycloaddition of two olefins to give a cyclobutane.”301 This analysis has several problems. For example, a MO analysis must involve the interaction of all filled bonding orbitals of the two ethylenes, taking into consideration the various perturbations that obtain with in-phase and out-of-phase interactions.
). The analogues to benzene/aromaticity and cyclobutadiene are made and discounted, as stated by Streitwieser,

“It should be emphasized that [the Diels-Alder transition state] is calculated as a modified benzene [Figure 38] but does not have benzene symmetry; the two π-systems to be joined lie in separate planes.”301
and
“Highly fluorinated double bonds… are particularly prone to [2+2] cycloaddition… In simple MO terms, [the transition state] becomes a distorted cyclobutadiene [Figure 38] this unsymmetrical addition is clearly analogous to that which may be involved in the Diels-Alder addition… In this connection, it is interesting that cycloadditions via quasi-eight-membered rings [ [4+4] or [6+2] ] are rare… it may be due to the electronic instability of the conjugated cyclooctatetraene structure.”301
-
Chapter 15 includes a discussion of the Cope and Claisen reactions. Streitwieser's representation of the “possible transition state for the Cope rearrangement of 1,5-hexadiene is shown by 23. The aromatic-like nature of this TS is evident.
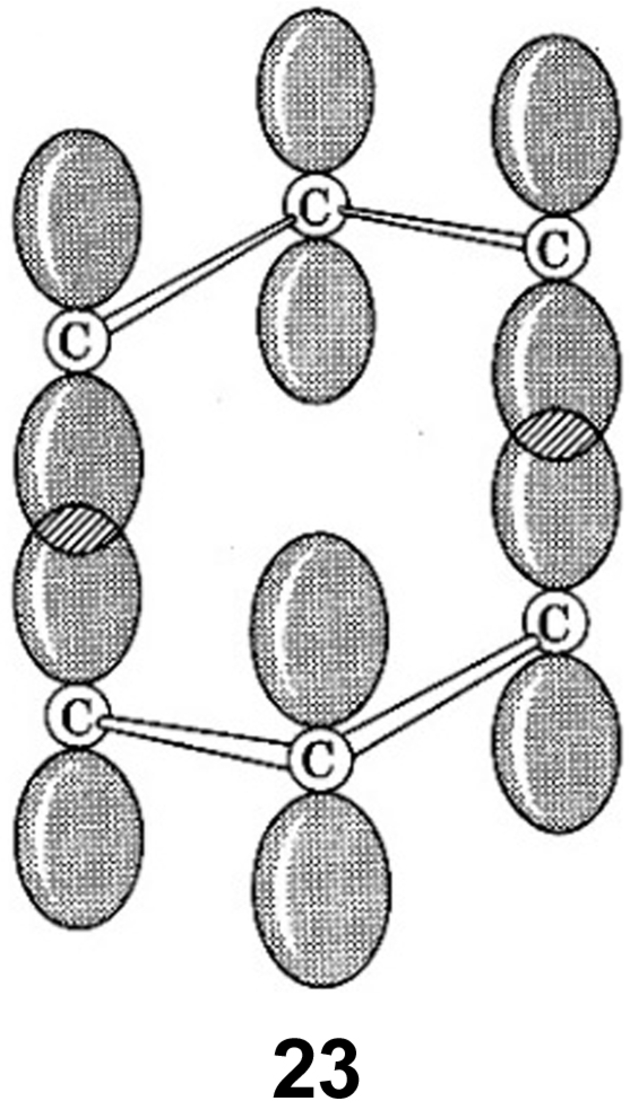
In a 2014 interview, on this matter, Streitwieser concluded,
“What I didn't do is relate these reactions to 4n and 4n+2 cycles. I could have but I didn't. I didn't make the jump… . but it was close.”312
3.16.4. Gateway: Aromaticity
According to Streitwieser, when writing his 1961 book,
“I did recognize that carbonium ions rearrange by way of a 2-electron cyclic TS and that carbanions do not because they involve 4 electrons and violates 4n+2 but it never occurred to me to put a node in the cycle and get a Möbius system.”313
and
“At that time [1960] I was aware that 2+2 cycloadditions (such as HI+HI=H2+I2) are disfavored because they are 4n and not 4n+2. In fact, I was tempted to put this type of argument in my MO book, but I didn′t because of fatigue. This was getting towards the end, and I wanted to finish it. I thought of including the ene reaction but there was not much literature on it at the time. But, although I was aware of the relationship between cycloadditions and 4n+2, I was not aware of con and disrotation and of the role of Möbius MOs in such systems. I also didn′t know enough about perturbation theory, but Dewar did and he of all people had the knowledge and ability to be competitive. A few years later, at the end of 1964, my focus of interest was carbanions and acidity. I wonder if I even noticed the Woodward-Hoffmann communication. I also at that time had other personal problems that were more important. My wife died in May of 1965 [a suicide] leaving me with two young children to include in my schedule.”314
3.16.5. Why and Why Not Streitwieser?
Why Streitwieser? He was an expert in MO theory, having written the most extensive book on the subject for organic chemists in 1961.192 He was an expert in aromaticity, in part because he had been a greaduate student with Doering in the early 1950s when Doering was devising Hückel's 4n 2 rule. He was both an experimentalist and a theoretician. He was a Berkeley colleague of W. G. Dauben who had discovered some of the no-mechanism clues in his laboratory. The two of them had talked about that chemistry.
Why not Streitwieser? I was able to interview him many times, twice in his home in Berkeley, once in San Francisco, and often by telephone and email. Let him speak for himself:
“At the time I wrote my MO book (and this is clear in the book itself) I was mostly concerned with the numerical energy aspects of MO theory and their possible correlation with quantitative experimental results. Much of my early research was concerned with that, such as measuring the acidity of methylarenes, aromatic substitution of polycyclic aromatic hydrocarbons, etc.; that is, to obtain experimental numbers for pi electron systems amenable to computation with HMO theory. (I also told this in my Adams Award lecture.)”315
Streitwieser refers to his theoretical studies that were typical for the pre-1965 era. These included reactivity indices which were being studied extensively by Michael Dewar and Kenichi Fukui. Streitwieser also said,
“Oosterhoff is not mentioned at all in my MO book. Considering the extensive literature studies I did in the late 1950s for the book, it suggests that he simply was not recognized as a theoretical organic chemist at that time. [And Oosterhoff's seminal suggestion appeared in the literature in January 1961,40 the year that Streitwieser's book was published.192] By contrast, Fukui, who was a physical theoretical chemist, contributed to theoretical organic chemistry and is cited a number of times in my book. But it's true that the book's discussion of cycloaddition reactions belongs to another [future] era. I knew that even at that time, as indicted by my call for further research, when I wrote, ‘Theoretical treatments [of cycloadditions] are rare and further research in this area should be encouraged.’.192”316
and
“I recall thinking I should talk to Bill Dauben about it but didn′t. Of course, I should have! How my life would have changed had I done so!”314
and
“As to why I, in particular, did not discover orbital symmetry myself, I think the answer is pretty simple: I just didn′t know enough. I knew enough MO theory to write a book but I didn′t really know it thoroughly enough; I didn′t really know it in my gut. My story in the preface of my book,
‘of the suspect, who, after long and diligent interrogation by experienced police officers, finally blurted out, ‘But fellows, I've told you more than I know already!’192
has a certain element of truth. I didn′t know enough about perturbation theory. I did learn it later, of course, and I believe my Science write up about the 1981 Nobel prizes317 is still as good an explanation of frontier orbital theory as one can find. But that came much later. Some aspects of perturbation theory I learned from Michael Dewar in his series of 1952 JACS papers, but I didn′t have enough background at that time to really understand them. I should, of course, have taken the time to learn perturbation theory more thoroughly at that time but I didn′t. There was always something else more pressing to do. In addition, there was also the language barrier between organic and physical chemists. We couldn′t explain to them what we needed to know, and they couldn′t teach us in language we could understand. I suspect this is true for many other organic chemists besides myself.”296
Streitwieser's story of the police questioning the suspect has several related antecedent perspectives. First a quote by one of the discoverers of transition state theory, Michael Polanyi.
“I shall reconsider human knowledge by starting from the fact that we can know more than we can tell. This fact seems obvious enough; but it is not easy to say exactly what it means.”318
And a related quote,
“Only the man who knows too little knows too much,” by Rex Stout, in a biography of fictional detective Nero Wolfe.319
3.17. Michael J. S. Dewar (1918–1997)320-323
3.17.1. Preliminary Comment
In 1965, Michael Dewar (Figure 39) clearly understood the breadth and scope of the no-mechanism reaction. In that year, he wrote,

Michael Dewar (left) with Paul S. Bartlett after his talk on π-complexes at a Bartlett group seminar, Harvard, 1951. Photograph courtesy J. D. Roberts.
“It is very difficult to devise experiments by which the mechanisms of reactions of this class can be established; they have for this reason been described, not altogether frivolously, as ‘no-mechanism’ reactions. Investigations along the lines indicated above could well resolve these problems. For this one would need extensive data for reactions involving aromatic and conjugated hydrocarbons of varying size and shape and free from extraneous substituents. Data of this kind are lacking, probably mainly because the need has not been recognized.”99
But the data were available and had been recognized by other chemists, as described at the top of this paper and in great detail in Paper 2 in this series.9 As discussed in Paper 16 in this series,324 Dewar was not always accurate in his pronouncements as to various historical facts in chemistry.
3.17.2. General Background Information
Like Woodward and Carl Djerassi, Dewar was a real “force of nature.” Roberts characterized Dewar as
“Peck's Bad Boy of chemistry. This man is in many ways a genius, broad of interests, quick of intelligence, very outspoken, and possessing a gargantuan humor. . . Dewar loves to take pot shots at anybody's sacred cows and then bursts into a sort of shy, nervous, embarrassed laughter. No statement seems too outrageous for him to make. Relatively short, built like one of those round-bottomed, regain-upright dolls, with a profile like New Hampshire's ‘Great Stone Face,’ in no time at all Dewar was a force to be reckoned with.”135
Dewar was born in India in 1918 where his father served in the India Civil Service. After schooling at Winchester College, Dewar completed his B.A., M.A., D.Phil., and a postdoctoral at Oxford with Robert Robinson. After six years (1945–1951) as a research chemist in physical chemistry with Courtaulds, he was called to the professorship of chemistry at Queen Mary College, University of London (1951–1959), to the University of Chicago (1959–1963), and then to the University of Texas at Austin (1963–1990). Among many honors and awards, Dewar was a Fellow of the Royal Society and a member of the National Academy of Sciences.
3.17.3. Gateway: Pericyclic Reactions
During the pre-1965 period of Dewar's career, he studied a considerable number of no-mechanism reactions. For example, while still a student in Oxford, in 1945, Dewar proposed
“in the initial hydrazobenzene salt, a non-localized π-electron migrates from ring 2 to ring 1 with consequent fission of the N−N bond to produce the complex molecule [Figure 40], composed of the aniline derivative A and the ion-radical B. Since the electron levels of the latter are incompletely filled, and since the π -orbitals of the rings will overlap, exchange forces should hold A and B together. The product will be called a π-complex. The ‘π–π’ bond in it will be of novel type, joining aromatic systems and not a pair of atoms.”325

Dewar's 1949 textbook Electronic Theory of Organic Chemistry2 discussed rearrangements some of which would later be termed pericyclic reactions. One, of course, was the Diels-Alder reaction. For Diels-Alder reactions having a dienophile activated by an electron withdrawing group, Dewar favored an ionic, multi-step reaction. For what he categorized as “non-polar Diels-Alder reactions,” Dewar criticized Meredith G. Evans's 1939 suggestion of an aromatic-like transition state327 on the basis of the non-planarity of the Diels-Alder transition state. Ironically, in 1966 and 1971 Dewar championed the Evans publication as a precedent for the Woodward-Hoffmann rules; this is discussed in detail in Paper 16) of this series.324 But in 1949, Dewar favored a diradical mechanism for the Diels-Alder reaction. In time, Dewar would evolve. For the Cope rearrangement (eq. 24), Dewar drew a complex of two allyl radicals though Dewar did not use that or any terminology. But Dewar, the champion of π-complexes,328 likely considered the dotted-lines in the middle structure of eq. 24 to be suggestive of a π-complex.
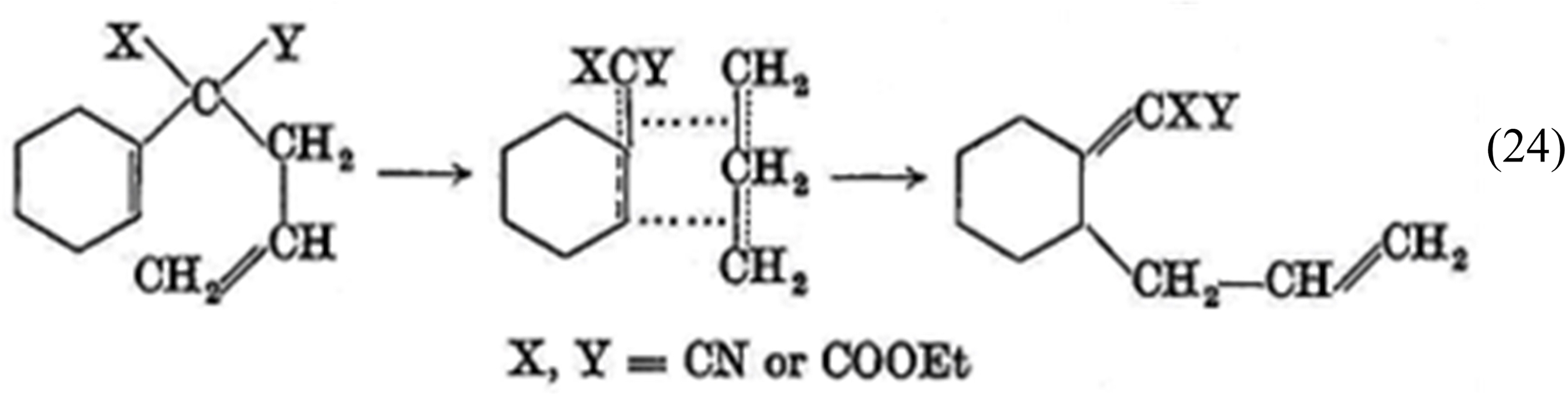
In 1951, Dewar discussed Reactions involving Cyclic Transition States.329 Dewar's interests in valence isomerizations and cycloadditions continued into the 1960s, with papers on the Diels-Alder reaction330 in which he engaged in a brief debate with Woodward on the mechanism of the Diels-Alder reaction331 and also studied the norcaradiene  1,3,5-cycloheptatriene valence isomerization.332
1,3,5-cycloheptatriene valence isomerization.332
In 1971, Dewar said in a review of his knowledge,
“certainly I have been including in my lecture course since the mid-fifties a discussion of what I then called ‘reactions involving cyclic transition states’, using as examples, among others, the Diels-Alder reaction, the Claisen rearrangement, the ene reaction, addition of diazomethane to olefins, and the pyrolysis of ricinoleic acid [c.f., the mechanism suggested by 24 which is a substructure of ricinoleic acid].”333
3.17.4. Gateway: Molecular Orbital Theory
In his 1985 oral history, Dewar said,
“What got me interested [in the 1940s] in theory as such was being asked by the Oxford University Press after the war to write a book on theoretical organic chemistry. When I started on it, I realized I didn′t know any quantum theory and I didn′t really know much physical chemistry. It took three years [for the book] to appear,2 because at that time, after the war, everything was a mess. But it had a whole lot of reviews, each worse than the last, and each time another review appeared the sales went up. It was read by a whole generation of young organic chemists, and it was the thing which really converted organic chemists to molecular orbital theory. It was the first account of organic chemistry in terms of molecular orbital theory.”323
In point of fact, there is hardly any molecular orbital theory and very few MO applications in Dewar's 1949 book.2 What did convert organic chemists to MO theory was the overwhelming success of the Woodward-Hoffmann rules 15 years later. But what the book actually did – or rather, what writing the book actually did – was to ramp up Dewar's own interest in the application of MO theory to organic chemistry. And the book surely taught Dewar the state-of-the-art of physical organic chemistry as of the mid-1940s. Dewar's first five years were already quite productive and demonstrative of an interest in a wide range of topics in organic chemistry (see above). His research career was about to explode in variety and magnitude.
Dewar used the Hückel method to calculate the resonance energies of the symmetrical allyl anion and radical and of benzene derivatives from heats of combustion (1946).334 He then modified the Hückel method to develop his first semi-empirical method and calculated the resonance energies of some pyrrole-type compounds (1946).334 In 1947, Dewar used Hückel MO theory to calculate the heats of formation of some π-complexes (Scheme 7).335 Also in 1947, together with the leading chemical theoretician of the era Charles A. Coulson, Dewar wrote a lengthy paper on Difficulties in the Application of the Molecular Orbital Theory to Mesomeric Ions.336 Coulson and Dewar concluded, in part,

Examples of some π-complexes studied by Dewar in 1947 using Hückel MO theory.335
“that the simple MO method as at present used, is semi-empirical in the sense that no a priori estimate can be made of the probable errors involved. Moreover it is known to fail badly in at least one case (light absorption) where a quantitative test is possible. Any extension of the calculations to new types of compound must therefore be experimentally checked before any confidence can be placed in their quantitative significance.”336
In his 1951 paper328 based on his 1950 lectures at the Montpellier Colloquium,337 Dewar illustrated his HOMO-LUMO interactions of silver salts with ethylene (Scheme 8). Note that Dewar included both HOMO(i)-LUMO(j) and HOMO(j)-LUMO(i), where i is the olefin/aromatic ring and j is the silver cation. It took 15 years before others caught up with the completeness of Dewar's treatment.338

Dewar formalized his perturbation theory which explained Scheme 8 chemistry in a series of six papers published in 1952.278, 339-343 In the first paper of the series,339 Dewar derived the “resonance energy” RRS of a complex RS (which could be the transition state of a reaction) composed of two interacting units, R and S (eq. 25). Notably, eq. 25 is essentially the same equation derived and used by Kenichi Fukui in several papers in the early 1950s344-347 and early 1960s,348 chronologically after Dewar, that ultimately was used by Fukui in his Nobel Prize-leading 1964 paper.348
Dewar had intense and continuing interest in this donor-acceptor picture of complexes328, 329 which led to very important donor-acceptor picture of metal complexes, namely the Dewar-Chatt-Duncanson model.349-351 In this model, Dewar clearly recognized the nodes/phases and symmetry aspects of molecular orbital theory in what may well be the first, and certainly one of the earliest, examples of a reaction governed by the overlap of molecular orbitals that interact in a bonding mode. Eq. 25 is notable for several reasons. It incorporates a frontier molecular orbital method as well as uses perturbation theory to calculate the stabilization energies due to molecular orbital mixing. Scheme 8 is notable because it focuses on certain HOMO-LUMO interactions that dominate all the other MO mixing interactions as the latter have larger energy separations (a larger denominator in eq. 25) and a greater orbital overlap (a larger nominator in eq. 25), thereby providing a valence bond-type graphic (Scheme 8) tending toward maximum delocalization, i. e., maximum stabilization.
In 1949, Dewar published a paper on A Modification of the Molecular Orbital Method: The LCMO Method in the journal Mathematical Proceedings of the Cambridge Philosophical Society. The SciFinder abstract of that paper is as follows:
“A modification of the molecular orbital method is described which leads to the same polynomial secular equation as does the linear combination of atomic orbital (LCAO) method, but via a simpler secular determinant. The basic idea is to divide the whole molecule into parts, calculate the molecular orbitals for these by the usual method, and then use linear combinations of these mol. orbitals as trial characteristic functions in a variational treatment of the whole molecule. The treatment is extended to heteroatomic systems. Examples of application are afforded by discussion of the treatment for benzyl, triphenylmethyl, and the 3 anthrylmethyls (9-, 1-, and 2-). A modification of the MO method is described which leads to the same polynomial secular equation as does the LCAO method, but via a simpler secular determinant. The basic idea is to divide the whole molecule into parts, calculate the MOs for these by the usual method, and then use linear combinations of these MOs as trial eigen-functions in a variational treatment of the whole molecule. Equations are developed for a number of cases, and it is shown that the computations of energy levels and MO′s from them is often much simpler than from the corresponding LCAO equations.”352
In 1950, Dewar published calculations “using the standard molecular orbital method,” presumably a Hückel MO calculation, on tropolone and found it to have “a resonance-energy comparable with that of benzene and probably somewhat greater.”353
In 1951, Dewar authored the chapter on Theoretical Organic Chemistry for the Annual Reports in the Progress of Chemistry.329 One sentence in that review distinguishes Dewar's interests from those of theoretical chemists of that era, that being an interest in chemical reactivity.
“The basic problem of theoretical organic chemistry is the prediction of the way in which a given change in structure (e. g., introduction of a substituent or replacement of carbon by a hetero-atom) will influence its reactivity.”329
Notably, Dewar did not include the role of stereochemistry in reactivity. But then, Derek Barton's discovery of conformational analysis had just been published,26 perhaps too soon for its impact to reach Dewar. That the review contained almost no use of MO theory to explain reactivity is indicative of the start of that art in 1951.
In 1952, together with H. Christopher Longuet-Higgins, Dewar published a paper entitled The Correspondence Between the Resonance and Molecular Orbital Theories354 in which the authors concluded,
“that a remarkable correspondence exists between the resonance theory and the molecular orbital method; and it is suggested that the resonance theory owes its success more to this correspondence than to the validity of its own premises.”354
Several of Dewar's pre-1950 publications utilize Hückel MO theory. Others enhance that approach while others make it clear that Dewar felt that that method was unsatisfactory for the current and future needs of chemistry. These papers, then, were harbingers of Dewar's six consecutive papers that appeared in the July 5, 1952, issue of the Journal of the American Chemical Society (pages 3341 to 3363, inclusive). These were all entitled A Molecular Orbital Theory of Organic Chemistry with the following subtitles:
Part I. General Principles339
Part II. The Structure of Mesomeric Systems278
Part III. Charge Displacements and Electromeric Substituents340
Part IV. Free Radicals341
Part V. Theories of Reactivity and the Relationship Between Them342
Part VI. Aromatic Substitution and Addition343
In these papers, Dewar introduced his perturbation theory, all highly mathematical treatments (see Figure 41).334

Page 3344 of Dewar's first publication of his perturbation theory, July 5, 1952.339 Reprinted with permission from: M. J. S. Dewar, “A Molecular Orbital Theory of Organic Chemistry. I. General Principles.” J. Am. Chem. Soc. 1952, 74, 3341–3345. Copyright 1952 American Chemical Society.
Into the 1950s and 1960s, Dewar continued to publish on the use of MO methods and the development of new MO methods.355-359 Ultimately, Dewar's group developed a number of semi-empirical methods such as MINDO, MNDO, and AM1, and he and others used them widely.
Dewar also knew group theory and the importance of molecular orbital symmetry (for orbital mixing and stabilization), and he applied this to 1,3-butadiene.360 His research infrequently centered around symmetry but he used symmetry arguments in his discussion of silver cation's interaction with ethylene (Scheme 8).329, 351
3.17.5. Gateway: Aromaticity and Antiaromaticity
In 1945, Dewar analyzed the data provided by Birkinshaw, Chambers and Raistrick in 1942 for the natural product stipitatic acid isolated from Penicillium stipitatum having the empirical formula C8H6O5 and proposed the then quite novel structures 26 and 27. One chemical property reported by Raistrick et al.361 for stipitatic acid was of particular importance to Dewar and to the history of chemistry. “Stipitatic acid is unchanged by bromine in acetic acid.”230 This observation is reminiscent of an exchange between noted detective Sherlock Holmes and Scotland Inspector Gregory in the short story Silver Blaze:
Inspector Gregory: “Is there any other point to which you would wish to draw my attention?”
Holmes: “ To the curious incident of the dog in the night-time.”
Inspector Gregory: “The dog did nothing in the night-time.”
Holmes: “That was the curious incident.”362
Dewar concluded,
“The stability of stipitatic acid, particularly to bromine, indicates the presence of an aromatic structure… If stipitatic acid actually has the resonating structures 25 or 26, it represents a new type of aromatic system; the parent cycloheptatrienolone might be termed ‘tropolone’. This system would be closely analogous to azulene… Cyclopentadienolone may prove to have an analogous structure.”230
Within weeks, Dewar published a second paper in which he almost solved a more important structure, that of colchicine; “more important” in that even today, colchicine has medicinal value. Again, Dewar called upon the tropolone ring to produce the colchicine structural proposal and explain a series of previously mysterious experimental observations during the structure studies by other chemists. Dewar proposed a regioisomer of colchicine, i. e., 28, but he was well aware of that possibility and not unreasonably considered that to be a lessor structural feature than the novel tropolone feature he had proposed. Dewar wrote,
“Of course, in [27] the methoxy and keto groups in ring C might be interchanged or the ring rotated about its junction with ring B; further degradation will be required to establish the exact orientation of the various substituents.”363
It is historically important to note that in 1934, George Barger and Otto Dorrer364 had suggested a structural relationship between stipitatic acid and colchicine in a paper that was cited by Dewar in 1945.363 Thus, Dewar was easily motivated to solve both structural problems simultaneously.
Dewar, in one motion, not only solved several structure determinations but initiated an entire new field of organic chemistry: nonbenzenoid aromaticity. As Woodward summarized Dewar's discovery of non-benzenoid aromaticity in his (Woodward's) 1965 paper on the total synthesis of colchicine,
“It was this construct which Dewar's imagination brought to light, and its recognition led directly to the development of a wholly new and fascinating chapter of organic chemistry.”365
However, Dewar's 1945 explanation was based on resonance theory. And his application of that considered both the 4n system and the 4n+2 system to be resonance stabilized (Scheme 9).

Dewar's 1945 explanation for the stability of stipitatic acid and prediction for stability of cyclopentadienolone, for which Dewar wrote, “Cyclopentadienolone may prove to have an analogous structure [as tropolone].”230
Dewar carried over his ideas regarding stability from stipitatic acid to colchicine. He wrote about colchicine that
“resonance with 28 account[s] for its stability; the system is analogous to γ-pyrone.”363
Forty years later, Dewar explained the source of his error (which he most certainly understood not that many years after making it),
“I knew so little about theory then [1945], that I interpreted it wrongly in terms of resonance theory. I didn't realize that this was actually Hückel's C7 aromatic system.”323
That is, in the 1940s, resonance theory as it was then understood, failed to explain the instability of cyclobutadiene.
Dewar's knowledge of Erich Hückel's research in the 1930s248, 250, 251, 279 did not improve Dewar's pedagogy in 1949 when his textbook Electronic Theory of Organic Chemistry2 appeared. Dewar's definition of an aromatic ring was:
“(1) Each ring-atom must have a p-orbital available for π-bond formation, and the p-orbitals must be so oriented that they can all overlap to form large π-orbitals.
(2) The total number of electrons in the p-orbitals must be the even number n or (n+1). . . cyclobutadiene should contain two unpaired electrons and so be a diradical; the failure to prepare it is not surprising.”2
Dewar published many papers pre-1966 on cyclobutadiene and thus was aware of antiaromaticity, or perhaps more accurately, was aware of the “negative stabilization energy” of cyclobutadiene,366 as early as 1952.99, 278, 354, 367-369
Dewar's six-part back-to-back series of papers278, 339-343 on A Molecular Orbital Theory of Organic Chemistry in the Journal of the American Chemical Society in 1952 was and is nearly incomprehensible (see Figure 41).337, 351 These papers embody Dewar's 1952 perturbation theory of quantum chemistry. In Part II of this series, in Theorem 33, Dewar wrote,
“Symmetrical monocyclic AH's [alternate hydrocarbons] with 4n conjugated atoms are biradicals. This follows from the method of proof of theorem 24; if the AH RS [“a mesomeric system”339 not otherwise defined], with 4n atoms is dissected into an odd AH R and one atom S, the NBMO of R survives in RS. Since NMMO's in an even AH can occur only in pairs (theorem 4), RS contains at least two NBMO's and is therefore a biradical. This theorem is not of much importance as cyclobutadiene does not seem to exist as a stable entity, and since the higher members of the series are prevented by stereochemical factors from adopting symmetrical coplanar structures [e. g., cyclooctatetraene].”278
This is, in Dewar's mind, his 1952 introduction of the concept of antiaromaticity.104 In 1971, Dewar wrote,
“. . . the term ‘antiaromatic’ was proposed [by Dewar] in 195214 on the basis of a simple extension of the Hückel MO method, using perturbation theory.”104
For reference 14 in the above quote, Dewar wrote,
“[14] M. J. S. Dewar, J. Amer. Chem. Soc. 74, 3341, 3345, 3350, 3353, 3355, 3357 (1952).”
Actually, the only discussion relevant to antiaromaticity that I could find in Dewar's 1952 papers is that reproduced immediately above, in the midst of Dewar's Theorem 33. In fact, I posit that the only way one could connect that excerpt from Dewar's 1952 papers to antiaromaticity is if one is determinedly looking for it with a quite flexible set of eyes and a fistful of imagination. Dewar's claim that this is the first public announcement of antiaromaticity is simply unwarranted.
Dewar's series of six papers was submitted on September 13, 1951. Doering's “2+4n” exposition of Hückel's rule was published in February 1951 and is not cited by Dewar. Nor does Dewar cite Erich Hückel's 1930’s papers on aromaticity which contained data quite relevant to Dewar's Theorem 33.
Dewar does point out the instability of cyclobutadiene several more times before he99 and Breslow98, 100 coin the term “antiaromaticity” in 1965. In 1958, Dewar said,
“Cyclobutadiene would have a negative resonance energy and I cannot encourage anyone to try to make it.”366
And in 1971 Dewar wrote,
“On a visit to Du Pont in 1958, when asked why certain olefins gave cyclobutene derivatives with dienes rather than ‘normal’ Diels-Alder products, I replied at once that the reactions must involve two step processes via intermediate biradicals since the concerted cycloaddition to a 4-ring would involve an antiaromatic transition state, while cyclisation of a biradical to a 4-ring rather than a 6-ring would be favoured from considerations of entropy.”333
3.17.6. Why and Why Not Dewar?
Why Dewar? By 1964 if not earlier, Dewar was aware of the no-mechanism problem. He was a speaker at the 1964 Reaction Mechanisms Conference at which Doering gave his lecture on the no-mechanism problem.82 He cited the no-mechanism problem in a book chapter that appeared in 1965,99 thus certainly written in 1964 or earlier.
Dewar was not just a nonconformist; he was a problem solver. He searched to correct the missteps of those before him and to reveal that which had confounded others. His interests were as broad as was chemistry and then beyond. His early research was experimental, and he continued having an experimental component in his research for two decades. His very first publication in 1941 was to correct certain structural missteps for yohimbine.370 Then followed his correct proposals for stipitatic acid (1945)230 and colchicine (1945)363, 371 and in 1948, he proposed “a new type of mol. linkage involving π-bonding.”372 He proposed a novel type of non-benzenoid aromaticity230 and thereby initiated a new field of chemistry. He became the first organic chemist – even more important, an organic chemist who was first an experimentalist – to use molecular orbital theory to explain chemical reactivity.
• Dewar was familiar with theory and experiment so that he could have come up with the principle of Conservation of Orbital Symmetry before Woodward and Hoffmann. Dewar
• Knew of Evans's application of aromaticity to the Diels-Alder reaction, a prototypical cycloaddition reaction;
• Had intimate knowledge of molecular orbital theory and especially discovered perturbation theory in 1952;
• Knew aromaticity, had discovered non-benzenoid aromaticity in 1945;
• By 1952, knew of and claimed he was the originator of antiaromaticity; and
• Had studied pericyclic reaction mechanisms.
Why not Michael Dewar? He was perfectly placed, with the knowledge and the experience.
• >One reason: All the other areas of chemistry, the competing stimuli that he was studying. In the 15 years from 1950 to 1964, Dewar published 172 papers covering a wide range of subjects. Included in Dewar's published interests were the following topics:
○ Electrophilic substitution
○ New heteroaromatic compounds
○ The tropylium ion
○ Acid-catalyzed rearrangements of alkyl aryl ethers
○ Chemical applications of nuclear quadrupole resonance spectroscopy
○ Resonance and conjugation
○ π-Complexes
○ Substituent effects
○ Electrophilic addition to olefins
○ Liquid crystals in solvents
○ Organometallic semiconductors
• Another reason, Dewar had no experience in steroid chemistry. Thus, his knowledge of the vitamin D chemistry was likely nil, thereby making it unlikely he knew of all those key chemical results pertinent to the no-mechanism problem.
• Dewar also had little interest in small ring chemistry and valence isomerizations other than that dealing with tropolones and 1,3,5-cycloheptatrienes. He may not have known of Vogel's39 and Criegee's16 mysterious reactions (eq. 5), for example. He also had little interest in photochemistry, so that aspect of alternating thermal – photochemical stereochemistries may have eluded him. As F. Thomas Bond recalled from his graduate school days with W. G. Dauben at the University of California, Berkeley, (who was heavily involved in vitamin D thermal and photochemistry),
“I also remember a Dauben group seminar attended by Michael Dewar of Chicago (summer to fall 1959).”373 “When Gerhard Fonken, one of WGDs brightest students talked with him (Dewar) about the photoisomerization of Vitamin D–the potential for understanding of the W−H rules was there but Fonken, like all of us at the time, did not know simple MO theory. My sense is that Dewar did not think this was very important!”374
Nor did Hoffmann in the first five months of his collaboration with Woodward think the problem offered to him by Woodward was very important.56 It was only after Hoffmann had heard a number of eminent chemists lecture on their puzzlement regarding no-mechanism reactions that Hoffmann began to appreciate the importance of the science.56
Indeed, Dewar has stated that the solution was obvious to him before W−H but the project did not seem so important.375
• Still another reason, Dewar was also writing a textbook Introduction to Modern Chemistry that was published in 1965,376 the Woodward-Hoffmann year. Dewar was mightily distracted.
• Dewar was a highly independent individual who often chose projects that were of particular interest to him, not necessarily of interest to the chemical community. Dewar was not deeply interested in the mechanisms of no-mechanism reactions even though he was aware of this phenomenon and did publish on the topic. In 1965, he published an essay in C&EN377 on Resonance, Conjugation and Hyperconjugation which preceded Breslow's C&EN article100 on Aromatic Character by more than six months (January 11, 1965 versus June 28, 1965). Dewar did not discuss antiaromaticity while Breslow did.
• Dewar's desire to explain a lot of chemistry using his a π-complex theory may well have kept him from seeing the benzidine rearrangement as a sigmatropic reaction.
• Dewar also was a strong opponent to frontier molecular orbital theory.99, 378 That being said, he used a FMO approach with appropriate molecular orbitals (with phases and nodes) to describe bonding (Scheme 8).
• Dewar's theoretical papers in the pre-Woodward-Hoffmann era were full of mathematical expressions and were difficult if not impossible (and uninspiring) for non-theoreticians to understand. See one page from Dewar's first paper in his new MO theory from 1952 (Figure 41).
• Zimmerman may have characterized Dewar accurately when he wrote,
“Mike could see many things as obvious in his mind but had not been published.”379
Dewar himself confirmed Zimmerman's explanation of his (Dewar's) behavior, saying
“All these ideas seemed to me obvious deductions from the literature and consequently not worth publishing.”333
That is, Dewar may well have underestimated the value of certain research results and associated explanations.
-
Zimmerman also said,
“I always felt a bit sorry for Mike Dewar. He didn′t get enough recognition. I don′t know but think he didn′t get awards. He was perhaps too bright for his time and didn′t apply himself to current problems and most people didn′t understand his theoretical arguments. They did understand the utility of his programming.”379
3.18. Authors of Important Pre-1965 Reviews: Jean Mathieu and Jacques Valls; Sara Jane Rhoads
Several individuals wrote review articles that gathered and organized the literature of no-mechanism reactions prior to 1965.
In 1957, Jean Mathieu and Jacques Valls at Roussel-Uclaf published Le tranfert électronique circulaire dans l′interprétation de certaines réactions de la Chimie organique [“Circular electronic transfer in the interpretation of certain reactions of organic chemistry”]. This 31-page review with over 700 references covered a very wide range of the literature of what was termed electrocyclizations, cycloadditions, and sigmatropic rearrangements by Woodward and Hoffmann in 1965 (Figure 42, top). A very short section presents mechanistic considerations, and one diagram offers a molecular orbital picture of the Diels-Alder reaction though without nodes/phases (Figure 42, bottom).

Two excerpts from the 1957 review of Mathieu and Valls.380
As mentioned above several times, in 1963, Rhoads published136 a 51-page review of the no-mechanism reaction including well over 110 references. Examples of electrocyclizations, cycloadditions, and sigmatropic rearrangements were included in Rhoads's review. Her paper also included a discussion of the Havinga and Schlatmann 1961 paper having the Oosterhoff suggestion of orbital symmetry control.40 But Rhoads did not mention Oosterhoff's suggestion nor discuss any aspect of quantum chemical control over the no-mechanism reactions.
Why did these authors not solve the pericyclic no-mechanism problem? For Mathieu and Valls, 1957 was too early. There were too few “laboratory curiosities”150 or otherwise inexplicable reactions by 1957 to recognize a mechanistic puzzle. The [2+2] versus the [4+2] cycloadditions could have been explained by a simple extension of Evans's 1939 paper327 in which the facility of the Diels-Alder reaction was related to the aromaticity of its transition state.
As for Rhoads, she is discussed in Section 3.7.
Of course, there were others who had written major reviews. In 1963, Vogel published a 10-page review on Valence Isomerizations in Compounds with Strained Rings with 71 references. Examples of electrocyclizations, cycloadditions, and sigmatropic rearrangements were included. In 1965, Seebach published a 10 page review on Strained Polycyclic Systems Consisting of Three- and Four-Membered Rings with over 50 references.381, 382 Vogel and Seebach are discussed in Paper 6 in this series.4
3.19. And There Were Still Others
There surely were many, many others. Too numerous to name, far too numerous to discuss. One that immediately reaches the mind: Albert Eschenmoser. He likely knew of Ranganathan's results (Figure 25), as his collaboration with Woodward on the total synthesis of vitamin B12 began in late 1964. Indeed, it was Eschenmoser who envisioned and used a remarkable photochemical sigmatropic rearrangement in his own synthesis of corrins in 1969.383 He was a master at reaction mechanisms. He had used the norcaradiene to cycloheptatriene valence isomerization as a key step in his 1959 epic total synthesis of colchicine.384 Why not Eschenmoser? Why not any of the others. I summarize the reasons in the next section.
4. Summary: Why Not the Others?
4.1. “It was in the air”
Many chemists were studying valence isomerizations, cycloadditions, a proton migration via cyclic transition states in the late 1950s and early 1960s. The discussions occurred in the literature, at scientific meetings, and in private conversations. Doering had labelled reactions like the Cope, Cope and Diels-Alder reactions as no-mechanism reactions,137, 138 and Sara Jane Rhoads had written a review article entitled Rearrangements Proceeding Through “No Mechanism Pathways.136
It is an interesting paradox: How could the mechanism of so many reactions in organic chemistry be so difficult to solve and yet be so easily solvable (or have the solution already in the literature, as with Oosterhoff40, 165 and Fukui348)? The next three subsections will offer some suggestions as to this paradox.
4.2. Individual Researcher's Internal Prerequisites
-
To go beyond their current and immediate research program, scientists must willingly reduce, to some extent, their current research interests or add resources to their current program. Research programs and their budgets are like zero sum games. Scientists are typically focused on their own on-going research programs, and there are practical, emotional, and even fiduciary reasons for research program continuity.
-
To solve a research problem, one must recognize the existence of that problem.
Chemists such as Doering and Rhoads, who certainly had a real grasp of the no-mechanism concept, had collected concerted reactions together as possessing similar chemical qualities, e. g., little if any rate variation with changes in solvent. But there was no suggestion in the literature before Woodward and Hoffmann that there would be a single, global mechanism that would obtain for all of these reactions.
-
Chemists were applying the up-to-then standard explanations for the no-mechanism reaction mechanisms. A search for a novel underlying explanation rooted in quantum chemistry was without precedent.
The 1950s and 1960s was the age of steric effects and conformational analyses. Every intelligent and up-to-date organic chemist since Odd Hassel, Barton, and Ernest L. Eliel would be expected to first analyze the situation using those concepts. Which is also what Woodward did, up to a point, until sufficient sets of alternating allowedness/forbiddenness coupled with stereospecificity was seen.
It is also true that a small number of chemists were using Hückel molecular orbital theory and simple versions of LCAO methods to correlate with chemical reactivity. But the chemical reactivities that were being studied were essentially reactivity indices, for example, estimates of electron densities at various carbon atoms in aromatic substitution reactions. Almost always, Hückel MO theory was applied to planar systems.
As Hoffmann related,
“Amazing, all those talented people, knowing, feeling that molecular orbital theory was important and held a key to understanding. But they were also stuck in π-electron theory, encouraged by the success of Hückel's rules. Trying to get out of the confines of the method – you can see that in Zimmerman's work, in Winstein and Simonetta – not quite finding their way to the extended Hückel method [which could treat three dimensional systems].”385
For most practicing organic chemists of the era, their knowledge of MO theory was nil. And the intuition to seek help from theoretical chemists had not yet evolved. Thus, Dauben did not seek help from his Berkeley colleague and MO theory expert Streitwieser or to his own brother Hyp Dauben who was also an expert in MO theory. And those few that did – Havinga going to Oosterhoff at Leiden; Doering going to Berry at Yale – they did not convert short conversations into serious, fruitful collaborations. As did Woodward and Hoffmann in 1964–1970.
-
Researchers must know the literature
Throughout this research, many instances were uncovered in which chemists are not citing relevant literature publications. The papers these excellent chemists missed were not hidden in obscure journals, and they were not in other disciplines. These are important papers in physical organic chemistry, published in leading journals by other well-known chemists. How can this happen? (1) Malice and other detrimental research practices?386, 387 Conscious disobedience?388 Neither. Interviews of chemists of that era finds no support for intentional misbehavior. (2) The literature was too big. It was big in the 1960s, and it lacked computerized search engines. However, that other chemists were finding those papers argues against this as a general explanation, though it might very well be true of Oosterhoff's suggestion in Havinga's 1961 and 1962 papers.40, 165 (3) Rushing to publish? Perhaps. (4) Unexacting? If so, it is (a) selective, and (b) likely to be a general quality of a scientist, exhibited in other circumstances.
But the observation is, many eminent chemists did not know all the relevant literature. My guess: selective reading of the literature.
4.3. Individual Researcher's External Prerequisites
-
Research resources: Computer software and hardware and students who were interested and able to write code and run programs
As Hoffmann summarized,
“An important actor that we haven't talked about is the emerging computer. That's what allowed me to set up and calculate three-dimensional molecules of the size of butadiene to phenanthrene.”385
-
Peter Chen et al. have pointed out that “For a time, it was almost a point of honor with both physical and organic chemists to profess ignorance of the other's field, and it remains a useful defense mechanism, if any is needed, to excuse the fact that specialization entails limitation as well as intensification of knowledge.”389
This consideration is symmetrical: it applied to both physical and organic chemists. One might wonder, was it more difficult for one or the other discipline to seek out and interact/collaborate with the other?
As Reinhard W. Hoffmann reflected,
“Looking back at the development of the Markovnikov free radical addition reactions, one cannot avoid noticing a decades-long induction period. Induction periods in chemical reactions may be caused by the (intended or coincidental) presence of an inhibitor. Are there material or immaterial inhibitors responsible for an induction period in the progress of science? Progress of science is fostered by a flux of information between individual scientists. Unfortunately, there are barriers, which inhibit such an exchange of information. Such barriers result, for example, from man-made compartmentalization of scientists, called specialization!”390
Few organic chemistry graduate students or postdoctorals had expertise or interest in writing software code and running programs that required manual input of chemical structures. Graphics terminals were still 20 years in the future.
This author remembers being the only organic chemist who used the sole computer connection in the entire Dyson Perrins Laboratory (the organic chemistry laboratory at the University of Oxford) in 1983–1984. Not until mid-1984 did I have access – for one structure – to a graphics terminal courtesy of Mike Mingos and R. Johnston in the Inorganic Chemistry Laboratory. But that was sufficient to certify that the pseudooctahedral organotransition metal complexes I was studying had been properly constructed by digital input.391, 392 Output was available only from the university's computer center via reams of extra-large sheets of computer paper. The Woodward-Hoffmann collaboration occurred two decades earlier.
-
Peer pressure and institutional requirements
Collaborations between theoretical chemists and experimental chemists were rare in the 1950s and 1960s. The style of top-ranked chemistry did not include such programs. Chemistry departments hiring physical chemists and chemical physicists were not favorably inclined to hire such individuals who were studying organic chemical reaction mechanisms. Hoffmann, who had offers from many top ranked universities in 1963 when he was considered a chemical physicist, found that those offers disappeared in 1965 when he was publishing extended Hückel calculations on organic chemical problems, e. g., on the structure and properties of hydrocarbons (1963),286 on the nonclassical ion (1964),393 on azine orbitals (1964).394 Even organic chemists were skeptical. Hoffmann had trouble publishing some of his pre-W−H studies in organic chemical journals, e. g., a submission on photochemical mechanisms (1964) was never published.395
4.4. Universal Prerequisites
4.4.1. Nationalistic trends in science
As Steve Weininger has discussed in detail, German organic chemists of the 1930s–1950s did not focus on physical organic chemistry for many reasons: a commitment to commercial applications of chemistry, therefore an emphasis on organic synthesis which was already a German chemistry tradition; and the position that theoretical or mechanistic research were not real science (which produces something tangible).37
Jeffrey A. Johnson recently produced a convincing argument that the development of quantum chemical-based research programs in Germany after World War II was hampered by modifications of educational programs in German universities due to Nazi influences.396
This disdain for theoretical chemistry in Germany is ironic, as Hoffmann commented,
“Why did theoretical chemistry and crystallography NOT flourish in the country that gave birth to them.”397
These factors altogether could explain why quantum chemical applications were diminished in Germany after World War II even though much of the initial theory was born there.
Also, in the 1950s and 1960s, interdisciplinary research was extremely uncommon in American and European academia.
Times were changing. In Germany, Criegee, Vogel, and Huisgen deserve much credit – the quality of their work made the reintegration of Germany into worldwide chemistry possible. It was not just their chemistry; it was their attractive, engaging, and persuasive personalities as well. With change of fashion toward the “new” synthetic organic chemistry of Woodward and Corey, the old synthetic chemistry of German chemists did not impress. It would take the next generation of German synthetic chemists like Dieter Seebach to arrive on the scene.
4.4.2. The negative effects of large research groups
Beginning in the 1960s, the growth of post-Sputnik governmental support for science, the expansion of the college and university footprint, the increase in research and development departments throughout the chemical and pharmaceutical industry, and the flood of chemistry students led to an explosive growth in the number of Ph.D. programs and graduate students in chemistry. This led most organic chemistry faculty at the elite graduate institutions to manage large research groups. This led many research groups to harvest more research results than many professors could or were willing to publish. That is, many professors published only a fraction of the research completed by their students. Woodward was one of the most famous examples. But there were others including my own Ph.D. advisor William G. Dauben. Another such individual discussed in Paper 6 of this series4 was Huisgen. But what was remarkable about Huisgen was that he published nearly 100 papers after his retirement.398 It is also likely that “large research groups” are less than optimum both for educational purposes, and for doing good science. It may be that some of the chemists discussed in these papers and others were too busy to see the clues and to discover the Woodward-Hoffmann rules.
5. Why does it Matter?
Choices dealing with the selection of research projects matter. They matter deeply. A Nobel Prize can depend on it.
One might speculate that it takes just as much time and energy to read the literature, conduct research, write and publish a paper on an average research project as on an exceptional one. To the extent that personal passion is related to project significance, project choice is critical to scientists. How would the history of chemistry changed had some “persuasive force” had directed either of the two Daubens or Berson, Dewar, Fukui, Havinga, or Zimmerman or any other chemist to work on the no-mechanism problem and nothing else?
The research of all those who did not discover orbital symmetry still mattered in the history of ideas and progress of science. They provided an increasing number of clues and mysterious reactions that ultimately pointed to the mechanistic solution or supported (or destroyed) the hypotheses presented. “Knowledge is communal; there are just so many ways of making it.” And for many of those discussed here, there were still tangible rewards for their contributions: cited in the Woodward-Hoffmann papers and subsequent reviews.
6. Conclusions
This series (a) examines the transformation of chemistry into the modern era through the lens of the history of the development of the W−H rules; (b) provides an intensive description of the history of the development of a breakthrough in science; and (c) discusses the role of individuals in the advancement of a field.
During the course of researching and writing the papers in this series, physical chemists have often said to me, “Orbital symmetry control is trivial. We all knew it way before Woodward and Hoffmann.” But this statement demands an explanation as to what the “it” in “We all knew ‘it’” means. Certainly, the physical and theoretical chemists knew the MOs of 1,3-butadiene and more complicated organic molecules. They also knew about correlation diagrams and how to apply them. They also knew about perturbation theory and how to apply it. But they did not know the organic chemistry; and they did not know the pericyclic no-mechanism problem. Because if they had, they would have published and ultimately received the Nobel Prize.
An entire discipline – organic chemistry – could not explain the stereospecificities and alternation in allowedness observed in pericyclic reactions until Woodward and Hoffmann's five communications were published in 1965. But that discipline – and the organic chemists – did not know enough MO theory or sufficiently understand the power of correlation diagrams or perturbation theory.
As Berson wrote,
“It is true that in several isolated instances, various authors prior to Woodward and Hoffmann had used essentially equivalent methods to explicate individual circumstances. In this connection, the work of Evans, who recognized the aromaticity of the Diels-Alder transition state, is especially noteworthy. An adaptation of Evans’ ideas to an interpretation of the transition state for the dimerization of olefins to cyclobutanes as antiaromatic was pointed out by Streitwieser, and Oosterhoff's correlation of thermal and photochemical transformations in the vitamin D series with the symmetry properties of the highest occupied molecular orbital now is well known.”399
This paper and the preceding two papers in this series3, 4 describe chemists – many of whom were among the elite of their discipline, recipients of the highest awards from their scientific communities – who did not publish the solution to the no-mechanism explanation. They, and there were many others who could be named, all together paint a clear picture of confusion before explanation. “We all knew it way before Woodward and Hoffmann” is simply not the reality of the events.
In his 1986 memoir of Adolf von Baeyer, Huisgen – one of the organic chemists whose contributions to the pre-1965 pericyclic no-mechanism problem are discussed in Paper 6 of this series4 – pointed out that
“Most historians commit a serious error in singling out only a few eminent personalities as participants in this intellectual struggle. ‘Time blots out small merit while fattening big glory,’ as the French writer Henri de Montherlant phrased the injustice.”400
These papers tell the research stories of a wide variety of personalities who possessed a diversity of individual academic circumstances, were funded by different mechanisms, and were joined by an even more diverse group of students, in institutions around the world. That so many chemists were publishing in the field of valence isomerizations, and cycloaddition indicates that, not one factor but many independent results came together into what eventually was recognized by Woodward and Hoffmann as a single mechanism. The more independent stories, the more realistic and complete is the picture.
That it was Woodward and Hoffmann, together with Oosterhoff and Fukui, with ideas from Berry and Heilbronner, and later contributions from Zimmerman and Dewar, and not any of the many others who were themselves brilliant scholars indicates that the leap in scholarship, this jump in the gap of knowledge, was deeply idiosyncratic. Each of the others could have solved the mechanistic problem. Interestingly, Kohler's generalization did not obtain:
“When expected rewards fail to materialize, it's tempting to blame someone other than yourself for the failure.”401
In these stories, most chemists took a rather pensive yet praiseful and admiring response to Woodward and Hoffmann's success. But not all chemists were praiseful and admiring of Woodward and Hoffmann, and it was not simply jealousy. Longuet-Higgins apparently felt that that he was not properly recognized for his discovery.402 And Corey certainly asserted his intellectual property rights.403-405 Corey's story and Longuet-Higgins's story will be published in Papers 13406 and 17,407 respectively, in this series.
Immediately following the W−H papers, chemists rushed ahead to use the quantum chemical tools newly revealed to them, further testing the W−H predictions and applying theory in new and expanded ways. The synergy of theory and experiment, a new themata in the words of Holton,408 was demonstrated and propelled by Woodward and Hoffmann. Organic chemistry as a field was advanced by successes achieved by many of the actors discussed in these papers, aided by the rapidly increasing power and versatility of computers.
Why was it Woodward and Hoffmann rather than anyone else? Why Woodward? Why Hoffmann? These questions will be answered in the next several paper in the series.
7. Coda
Jerome Berson may well have provided the best summary when he wrote,
“Our question about how so many sophisticated chemists missed the orbital symmetry concept has made me think more about just what was the big breakthrough. In my view, it was the recognition that the phase properties of the reacting orbitals (somewhat misleadingly called “orbital symmetry” by W−H) control the reaction. This crucial property was not recognized by most chemists in the immediately preceding period. For example, in his review article on nucleophilic substitution,299 Streitwieser shows a picture that represents the orbital overlaps in the linear transition state of the SN2 inversion reaction, but he doesn′t explain why that arrangement is better than a bent, retention transition state. Similarly, Breslow's book of about the same time gives the reason why inversion is preferred as ‘better bonding in the transition state.’ But what is “better′ about it he doesn′t say. The answer appeared a few years later in, I believe, Salem's use of Fukui's frontier orbital theory, and also in a footnote in a paper by myself and Bob Bergman, using the W−H notation. The key insight that most chemists missed was that the orbitals′ phases or signs change after a node is encountered. That is what Oosterhoff was getting at, and it was at the base of Heilbronner's formulation of Möbius conjugated systems, and what Zimmerman used in his formulation of Hückel vs Möbius transition states. It was also the basis of W−H hypothesis of suprafacial vs antarafacial orbital pairing and of course of Longuet-Higgins's alternative formulation using orbital correlation diagrams. All of these use the phases as key elements.”243
8. Coda to a Coda
The first sentence in Frank H. Westheimer's 1960 memoir on Morris Kharasch for the National Academy of Sciences is,
“THE TRANSFORMATIONS of organic chemistry can be roughly divided into two categories: polar reactions and free radical reactions.”409 [Emphasis in original.]
What about valence isomerizations, tautomerizations,8 and concerted reactions? This is the same Westheimer who, by 1960, had already published the Kirkwood-Westheimer principle of electrostatic influence on organic reactions (1938);410, 411 had published his force field calculations on racemization of optically active biphenyls (1946);412 had published his research on enzymatic transfer of hydrogen from alcohol to DPN (1951);413 and was just a year from writing his major review on primary kinetic isotope effects (1961).414 This was a researcher who had wide experience in physical organic chemistry. One could claim that Westheimer's sentence just reflected his own research interests. But clearly, Westheimer had extremely wide interests. Was it that a large swath of organic chemists was simply not interested in concerted (pericyclic) reactions at that time? Or were they just focused on their own research projects. I believe the latter.
Acknowledgements
I thank the following deceased chemists who provided helpful discussions over the years: Douglas Applequist, Derek H. R. Barton, Jerome Berson, William G. Dauben, William von E. Doering, Charles H. DePuy, Gerhard J. Fonken, Rolf Huisgen, Subramania Ranganathan, John D. Roberts, Andrew Streitwieser, and Howard E. Zimmerman. I also thank Mrs. Douglas Applequist, Celia Arnaud, E. J. Corey, Albert Eschenmoser, Richard K. Hill, Alfred G. Hortmann, Reinhard W. Hoffmann, Joseph Lambert, Maitland Jones, Jean-Marie Lehn, Charles L. Perrin, Dieter Seebach, Patrick Shea, Steven M. Weinreb, several anonymous readers of an earlier version of this paper, the reviewers of this paper, and most especially Roald Hoffmann for helpful comments and discussions. I thank all those who have provided photographs, as acknowledged in the text. I thank the staff of the Boatwright Memorial Library of the University of Richmond for their continued technical support, and the archives at the Science History Institute and Harvard University for the reproduction of certain items.

