

9,10-Anthraquinone hinders β-aggregation: How does a small molecule interfere with Aβ-peptide amyloid fibrillation?
Abstract
Amyloid aggregation is linked to a number of neurodegenerative syndromes, the most prevalent one being Alzheimer's disease. In this pathology, the β-amyloid peptides (Aβ) aggregate into oligomers, protofibrils, and fibrils and eventually into plaques, which constitute the characteristic hallmark of Alzheimer's disease. Several low-molecular-weight compounds able to impair the Aβ aggregation process have been recently discovered; yet, a detailed description of their interactions with oligomers and fibrils is hitherto missing. Here, molecular dynamics simulations are used to investigate the influence of two relatively similar tricyclic, planar compounds, that is, 9, 10-anthraquinone (AQ) and anthracene (AC), on the early phase of the aggregation of the Aβ heptapeptide segment H14QKLVFF20, the hydrophobic stretch that promotes the Aβ self-assembly. The simulations show that AQ interferes with β-sheet formation more than AC. In particular, AQ intercalates into the β-sheet because polar interactions between the compound and the peptide backbone destabilize the interstrand hydrogen bonds, thereby favoring disorder. The thioflavin T-binding assay indicates that AQ, but not AC, sensibly reduces the amount of aggregated Aβ1–40 peptide. Taken together, the in silico and in vitro results provide evidence that structural perturbations by AQ can remarkably affect ordered oligomerization. Moreover, the simulations shed light at the atomic level on the interactions between AQ and Aβ oligomers, providing useful insights for the design of small-molecule inhibitors of aggregation with therapeutic potential in Alzheimer's disease.
Introduction
Fibrillar aggregation and plaques deposition of the β-amyloid peptide (Aβ) in the brain are common hallmarks of Alzheimer's disease. The Aβ peptide is a 39- to 43-residue segment generated by proteolysis of the amyloid precursor protein. Although little is known on the link between the aggregation mechanism and neurotoxicity,1 experimental evidence indicates that soluble oligomers and fibrillar precursors of Aβ may be the neurotoxic species.2
Several therapeutic strategies have been suggested for blocking different key-steps in the amyloid aggregation process, including the direct inhibition of the aggregation by using either peptides or small molecules.3, 4 N-methylated peptides5, 6 and L/D-polypeptides7 were shown to block Aβ aggregation. Several nonpeptidic low-molecular-weight molecules were observed to reduce Aβ fibril formation and cytotoxicity,8-10 in particular, scaffolds with aromatic or heteroaromatic rings have been identified as potent inhibitors of amyloid aggregation.11 As an example, indole derivatives inhibited fibril formation of Aβ peptide12 and lysozyme.13 Rifampicin and p-benzoquinone reduced the toxicity of islet amyloid peptide aggregates14 as well as inhibited amyloid fibril formation of hen egg-white lysozymes.15 Anthraquinones were shown to be effective inhibitors of tau protein aggregation.16 Notably, some small molecules active in vitro also showed beneficial activity against Alzheimer's disease in a mouse model.17
Molecular dynamics (MD) simulations have shed light on the very early events of amyloid aggregation.18-28 These studies have focused on the driving forces governing the β assembly, emphasizing the role of aromatic packing,19 hydrophobic forces20, 21, 23, 27 and electrostatic interactions.20 The arrangement of peptides within oligomers and the process of reorganization have also been investigated by MD studies.18, 19, 22, 24, 26 Moreover, the crucial role of amino acid sequence in determining the propensity to form aggregates has been studied in detail.23, 29 Recently, based on simulations of the inhibition of Aβ16–22 fibrillation by a N-methylated peptide, it has been postulated that the inhibitor can bind to different sites of a preformed fibril, and thereby perturb it via different mechanisms.30 Because of both poor oral absorption and low penetration through the blood-brain barrier, peptidic inhibitors have much smaller potential as drugs than small nonpeptidic compounds, although important exceptions based on nonnatural aminoacids exist.31 The mechanism of inhibition of fibril formation by small compounds is however still obscure.
Here, we analyze the influence of two planar and tricyclic compounds on the early phase of ordered aggregation of a segment of the Aβ peptide that promotes oligomerization. Implicit solvent MD simulations are used to investigate the aggregation of three Aβ14–20 (Ac-H14QKLVFF20-NHMe) terminally blocked heptapeptides in the presence and absence of AQ or AC. The Aβ14–20 segment is chosen because it has a high β-aggregation propensity according to biophysical experiments,32, 33 as well as atomistic simulations29, 34 and a phenomenological equation based on physicochemical properties of the primary structure.35 Notably, the MD simulations show that a small substitution in molecular structure (two carbonyl groups replacing two aromatic CH) results in significant differences in the ability to influence early aggregation. The simulation results are validated by the thioflavin T (ThT)-binding fluorimetric assay.
Results and Discussion
Properties of Aβ14–20 aggregates with and without tricyclic compounds
During the simulation the three-peptide system explores many different configurations, including aggregated, disaggregated, β structures with a variety of registers, and other spatial arrangements. The P2 order parameter (described in the Methods section) has been adopted to monitor the degree of orientational order within the oligomers: a value close to one corresponds to an ordered trimer, with either parallel or antiparallel β-sheet, while a value close to zero reflects a fully disordered system [Fig. 1(A)]. The frequency histograms of P2 for the unperturbed and perturbed systems [Fig. 1(B)] display a prominent peak at P2 = 0.8, and a shoulder for P2 values lower than 0.5, which includes disordered aggregates and isolated peptides. The P2 value for which the three distributions cross each other (P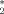 = 0.665) is chosen as the crossover value between ordered and disordered states. With this definition, the values of order-disorder ratio r can be calculated (Table I). AQ perturbs the oligomer order more than AC, suggesting that the quinonic moiety significantly contributes to the process of disorganization.
= 0.665) is chosen as the crossover value between ordered and disordered states. With this definition, the values of order-disorder ratio r can be calculated (Table I). AQ perturbs the oligomer order more than AC, suggesting that the quinonic moiety significantly contributes to the process of disorganization.

AQ hinders β-sheet formation more than AC. (A) Time series of the nematic order parameter P2 for the free system (black), and in presence of AC (red) or AQ (blue). A similar behavior was observed in the other nine runs for each system. The magenta arrow indicates the temporal location of the snapshots shown in Figure 2. Molecular structures of AC and AQ are represented in the insets. (B) Distribution of the nematic order parameter P2. Values of P2 close to 0.2 and 0.8 correspond to disordered conformations and β-sheet structures, respectively. The three distributions cross each other at P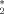 = 0.665 (vertical solid line), which is the threshold value that separates the ordered from the disordered phase. (C) Interpeptide interaction energy distributions (solid lines, left y-axis; see Methods section) and average P2 as a function of interaction energy (black circles, right y-axis). The two peaks of the energy distributions correspond to a peptide in the centre of an ordered oligomer (about −80 kcal/mol) and a peptide at the edge of an ordered oligomer (about −40 kcal/mol). The shoulder of the energy distribution at values of about −20 kcal/mol contains events with disordered or partially ordered oligomers. The P2 values higher than 0.5 for interaction energy close to zero is due to the propensity of the isolated peptide for an extended conformation. The insets in (B) and (C) are schematic pictures of the oligomer conformations. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
= 0.665 (vertical solid line), which is the threshold value that separates the ordered from the disordered phase. (C) Interpeptide interaction energy distributions (solid lines, left y-axis; see Methods section) and average P2 as a function of interaction energy (black circles, right y-axis). The two peaks of the energy distributions correspond to a peptide in the centre of an ordered oligomer (about −80 kcal/mol) and a peptide at the edge of an ordered oligomer (about −40 kcal/mol). The shoulder of the energy distribution at values of about −20 kcal/mol contains events with disordered or partially ordered oligomers. The P2 values higher than 0.5 for interaction energy close to zero is due to the propensity of the isolated peptide for an extended conformation. The insets in (B) and (C) are schematic pictures of the oligomer conformations. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
| System | ra | Activityb (%) | Aromatic contactsc (no. contacts per frame) |
|---|---|---|---|
| Unperturbed | 10.2 | ||
| with AC | 9.9 | 11 | 0.262 |
| with AQ | 6.7 | 33 | 0.242 |
- a Ratio between order and disorder events sampled in the trajectories and estimated by Eq. (1).
- b Inhibition of fibril formation measured by ThT fluorescence at a concentration of 30 μM Aβ1–40 and 100 μM AC or AQ.
- c Average number of aromatic contacts between the compounds and the phenyl ring of phenylalanine normalized by the total number of frames of simulations.
The frequency distribution of interpeptide interaction energies [Fig. 1(C)] shows two peaks. By visual inspection, and by comparing the energies with the average P2 [circles in Fig. 1(C)], we could assign the oligomer structures relative to the different regions in the energy [see insets in Fig. 1(C)]. The peak at −80 kcal/mol and the peak at −40 kcal/mol correspond to a peptide placed in the centre and at the edge of an ordered trimer, respectively. From the plot it is evident that the simulations with AQ have more events with interaction energy close to zero, originating from unstructured peptides bound to the oligomeric or isolated monomers. This indicates that the system's order is perturbed by AQ, which is able to intercalate into the oligomer and influence its structure.
Binding mechanism
The interactions at the basis of the activity of AQ are the hydrogen bonds, the aromatic contacts and, moreover, the ability to establish a favorable interaction between the central electron-poor quinonic ring and the electron-rich peptidic carbonyls, which here is called as π+δ− interaction (see Figs. 2 and 3). A typical series of events leading to β structure disruption by AQ is described in Figure 2. Once approached the oligomer, AQ separates the ordered peptides by interacting with amide hydrogens (blue dashed lines). It then penetrates into the oligomer, interfacing the carbonyl oxygens (red dashed lines). The β-sheet disruption is very rapid (less than 1 ns), which is in part a consequence of the low friction constant.

Atomistic details of β-sheet disruption by AQ. The times of the snapshots correspond to the time interval emphasized by an arrow in Figure 1(A). The quinonic moiety interacts with the peptide oligomer in a kind of “butter-knife” mechanism. AQ approaches the ordered oligomer (A) and starts to interact via hydrogen bond interactions (B and C, blue dashed lines). It subsequently intercalates into the ordered structure via hydrogen bonds and π+δ− interactions (D–F, red dashed line) and causes the disruption of the oligomer (F). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]

Interactions between backbone polar groups and AQ or AC. (Top) Positions of backbone carbonyl oxygens and amide hydrogens that are within 5 Å from any atom in AQ or AC are shown by dots to display the corresponding coordination. Denser clouds indicate a volume with higher coordination. (Bottom) π+δ− interaction scheme. Two electron-rich (δ−) peptidic carbonyl oxygens interact with both faces of the electron-poor (π+) AQ central ring.
The distribution of backbone oxygens (see Fig. 3, top) shows the enrichment of coordinated carbonyl oxygens around the quinonic moieties of AQ with respect to the condensed aromatic rings of AC. Being able to coordinate two amide hydrogens, and two backbone carbonyl oxygens at the same time, the quinonic moiety plays a major role in the intercalation into the oligomer. In this way, AQ is able to sequestrate not only donors, but also acceptors of interchain hydrogen bonds, which has three main consequences: (1) the disruption of the local intermolecular hydrogen bond geometry, (2) destabilization of the β-sheet structure, and (3) global disorganization of the oligomer, that is, reduction of the total order. On the other hand, AC is not able to interact with the peptides by hydrogen-bonds, or π+δ− interactions. Hydrophobic interactions alone are apparently not sufficient to perturb the ordered β architecture, as indicated by the similar values of r of the unperturbed and AC systems.
The number of aromatic contacts between the compounds and the phenyl rings of phenylalanine is reported in Table I. This interaction is slightly less frequent for AQ. This is due to the fact that, if compared with AC, AQ has also the ability to interact with peptide polar moieties, which are competitive with the hydrophobic ones. These results suggest that the aromatic interactions alone are not sufficient to drive the perturbation of Aβ14–20 ordered oligomers, although they may favor the encounter of the tricyclic molecule with the peptides.
Experimental results on Aβ1–40
The anti-aggregating activity of AC and AQ was determined in co-incubation experiments with Aβ1–40 by monitoring maximal ThT emission intensity over the course of 21 days (see Methods section). As shown in Figure 4, the amyloid aggregation obeys the characteristic nucleation-dependent pattern, with three distinct phases: initial nucleation (lag phase), elongation, and equilibration. Under the present experimental conditions, the system reached its final equilibration phase after about 20 days. The shape of the curves presented in Figure 4 indicates that the final plateau is influenced by the presence of AQ and much less by AC. The ThT fluorescence emission intensity in the co-incubation with AQ (blue symbols) was nearly 33% lower than with the control peptide alone (black symbols). A significantly lower inhibitory effect, 11% decrease of ThT fluorescence emission intensity at the final plateau, was observed for AC (red symbols). The different inhibitory effects of AC and AQ are consistent with the MD simulation results (Table I). A quantitative agreement is not expected because of differences in the sequence (full-length Aβ in ThT assay versus seven-residue segment in MD), in relative mass concentrations, and temperature values (see Methods section).

Time-dependent aggregation of Aβ1–40 30 μM, incubated in PBS (phosphate 10 mM, NaCl 100 mM, pH 7.4) at 25°C and monitored by ThT fluorescence. Error bars are standard deviations calculated from triplicate measures.
The elongation rate marginally decreases in the presence of AQ, while the lag phase is not influenced at all by AQ or AC. Yamada and coworkers showed that nordihydroguaiaretic acid, curcumin, and rosmarinic acid, though reducing the amount of fibril at the equilibrium, did not extend the length of the lag phase in the formation of fibrillar Aβ, nor the time to proceed to equilibrium.36 Recently, to explain the variable influence of compounds on Aβ42 aggregation kinetics, a new mechanism of inhibition was suggested.37 Compounds able to influence the elongation and the steady-state, but not the lag-phase, can prevent the self-assembly by blocking only the interstrand hydrogen-bond formation, in agreement with the AQ-binding mechanism discussed earlier, and not by stabilizing the nonamyloidogenic conformations of the polypeptide.
ThT measurements alone cannot unambiguously distinguish between inhibition of amyloid aggregation and competition of compounds with ThT binding. Meng et al.38 showed that it is possible to determine whether an inhibitor is a false positive by adding the compound to a preincubated fibril sample and measuring ThT fluorescence thereafter. A fast decay (within few minutes) of the signal indicates that the compound is indeed competing with ThT. We measured the ThT fluorescence just after adding AQ and AC to two preincubated Aβ samples. Within few hours, we couldn't observe any signal decrease (data not shown), allowing us to exclude any direct competition between AC/AQ and ThT at the ThT-binding site.
Abbreviations: Aβ, β-amyloid peptide; AC, anthracene; AQ, 9,10-anthraquinone; MD, molecular dynamics; ThT, thioflavin T.
Methods
Simulation protocol and analysis
The MD simulations were performed with the CHARMM program.39 The peptide and compounds were modeled using the united atoms CHARMM PARAM19 force field with its default truncation scheme for nonbonding interactions (cutoff of 7.5 Å). Hydration effects were accounted for by using SASA, a solvent-accessible surface based implicit model.40 Partial charges for AQ were computed with the modified partial equalization of orbital electronegativity (MPEOE) algorithm.41, 42 To be consistent with the united atom model, charge summation of carbon atoms and their directly connected hydrogens was performed. The results were in agreement with the original CHARMM PARAM19 partial charges for phenylalanine, tryptophan and peptidic carbonyl. The difference in electronegativity between the oxygen atom in the peptidic moiety (partial charge: −0.55) and in the quinonic central ring (partial charge: −0.51) is due to the fact that the peptidic carbonyl is directly linked to a nitrogen, more electron-withdrawing with respect to the carbon atoms in the quinonic ring. There is a favorable interaction energy of −3.6 kcal/mol between N-methylacetamide and p-benzoquinone, computed at low dielectric conditions (i.e., distance depending dielectric function, without the SASA solvation contribution). This value is in agreement with the −3.9 kcal/mol value obtained by ab initio QM calculations.43 To validate the parameterization of the bonding energy terms, two short MD simulations were performed for AQ and AC, using the SASA solvation model. Both molecules are stable, and the average bond distances and bond angles are comparable with the crystallographic data.44
The simulation box was prepared by introducing three monodispersed replicas of the same heptapeptide, with or without the presence of a single small molecule (AQ or AC). The concentration ratio peptide:compound of 3:1 is the minimal choice to have inhibitory effects and complex oligomeric structures. Note that the mass ratio peptide:compound is about 16 for the simulation and seven for the experiments (see ThT-Binding Assay section). To have the same mass ratio, one would have either to halve the compound concentration in the ThT assay, or to double the number of compounds in the simulation. In the first case the inhibition effect of AC would be undetectable. In the second case the compound-compound interactions would generate undesirable spurious effects, which is a disadvantage if one is interested in the oligomer–compound interaction mechanism. Simulations were carried out with periodic boundary conditions at fixed peptide concentration of 4.88 mM (the simulation box side was set to 98 Å), using Langevin integrator at low friction constant (0.1 ps−1) and at the temperature of 330 K, which yields reversible aggregation within a reasonable computational time. Ten, 2.5 μs long, independent simulations were run for each system. A 2.5-μs run takes 3 weeks on a single AMD Opteron 252 CPU at 2.6 GHz.

The unit vector 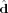 that defines a preferential direction is the eigenvector of the order matrix that corresponds to the largest positive eigenvalue. The N molecular unit vectors
that defines a preferential direction is the eigenvector of the order matrix that corresponds to the largest positive eigenvalue. The N molecular unit vectors 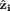 are built joining the Cα atom of residue i to the Cα atom of residue i + 2 (N = 3 × 7). The values of P2 ranges from 0 to 1, which correspond to complete disorder and complete order, respectively. The complete order is achieved when all the unit vectors are parallel or antiparallel, while the disorder is obtained when none of unit vectors is parallel to any of the others.
are built joining the Cα atom of residue i to the Cα atom of residue i + 2 (N = 3 × 7). The values of P2 ranges from 0 to 1, which correspond to complete disorder and complete order, respectively. The complete order is achieved when all the unit vectors are parallel or antiparallel, while the disorder is obtained when none of unit vectors is parallel to any of the others.
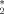 is a value of the order parameter chosen such that it separates the ordered from the disordered phase. Thus, the order-disorder ratio r is defined by the number of events where the system has a nematic order parameter lower than P
is a value of the order parameter chosen such that it separates the ordered from the disordered phase. Thus, the order-disorder ratio r is defined by the number of events where the system has a nematic order parameter lower than P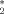 (disorder) and greater that P
(disorder) and greater that P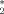 (order):
(order):
 ()
()Furthermore, the activity of the compounds is measured by calculating the interpeptide interaction energy, which is the CHARMM nonbond energy (van der Waals plus electrostatics) of a single peptide with the other two, without considering the compound molecule.
Aromatic contacts between the tricyclic compounds and the phenyl ring of Phe are calculated as follows. Given a trajectory frame, the centroids of the three rings of AC or AQ are calculated, together with the centroids of the six phenyl rings of Phe belonging to the three peptides. If any compound centroid is within 4 Å from any Phe centroid, then an aromatic contact is accounted. Note that more than one aromatic contact can be present at the same time, because the compounds have two sides available for the interfacing. The AC/AQ ratio of aromatic contacts is robust with respect to the choice of the cutoff up to 5 Å.
ThT-binding assay
The traditional spectrofluorimetric assay with ThT was chosen to assess the influence of the two tricyclic molecules, AQ and AC, on the kinetic of aggregation of Aβ1–40. This technique easily enables to monitor the formation of β-sheet structures of aggregating peptides with ThT that leads to a marked increase of the fluorescence of ThT at defined wavelengths. Nevertheless, the β-sheet/ThT interaction is unselective and does not discriminate among oligomers, protofibrils, and other transient species formed across the aggregation pathway.
The spectrofluorimetric assay was performed through the classical method of LeVine.45 Briefly, ThT solution for fluorescence experiments was 25 μM in 25 mM phosphate buffer (pH 6.0). Quartz cuvette for aggregation tests contained Aβ1–40 30 μM control peptide (from AnaSpec, San José, CA) alone, or added either with AC or AQ 100 μM, in phosphate buffered saline (phosphate 10 mM, NaCl 100 mM, pH 7.4) containing 20% dimethyl sulphoxide to solubilize the test compounds. The concentration of compounds was chosen to have the highest yield of inhibition without precipitation effects. Samples were incubated at 25°C for 3 weeks. During this period, spectrofluorimetric readings were made in triplicate by dilution of 30 μL of sample in 470 μL of ThT solution at 25°C. Analyses were performed with a Perkin-Elmer LS 55 fluorimeter, in a 700-μL quartz cuvette, using FLWinLab software. Parameters were fixed as follows: excitation wavelength at 440 nm with 5-nm slit; emission at 485 nm with 10-nm slit; integration time 2 s. Plots of fluorescence emission intensity versus time (see Fig. 4) showed the typical sigmoidal curve of aggregation kinetics. Activities were calculated as percent of inhibition of free peptide aggregation after 3 weeks.
Conclusions
In previous works, the Aβ14–20 heptapeptide was shown to have high aggregation propensity by biophysical experiments32, 33 and computational approaches,34, 35 suggesting a key role in the self-assembly of the full-length Aβ peptide. We have therefore hypothesized that the perturbation of the aggregation propensity of Aβ14–20 can influence the assembly properties of the full-length Aβ sequence. Here, implicit solvent MD simulations have been used to investigate the effect of AQ and AC on the β-aggregation of three replicas of Aβ14–20. The nematic order parameter P2 was used to monitor the perturbation extent of β-aggregation.34 Compared with AC, which is almost inert, AQ destabilizes the interstrand hydrogen bonds through favorable polar interactions with the backbone of the peptide. The quinonic moiety is able to tightly bind peptide backbone carbonyl oxygens and amide hydrogens through π+δ− interactions and hydrogen bonds, respectively, facilitating the intercalation of the molecule into the oligomer. The stability of the π+δ− interaction46 has been previously documented in ab initio QM calculations,43 the crystal packing of isocyanurate derivatives resolved by X-ray diffractometry,47 and coordination of anions with electron-poor rings.48, 49
Aromatic interactions have been shown to be determinant in the inhibition of fibril formation as well as the reduction of amyloid toxicity.11 Our simulations reveal that such interactions favor the formation of the compound–oligomer complex, but are not sufficiently strong to significantly perturb the β-sheet formation of Aβ14–20, as observed by comparing AC with AQ.
Perturbation of multimeric assembly is the basis for the activity of small compounds able to inhibit amyloid fibril formation. AQ only marginally affects the equilibrium properties of the unperturbed oligomeric system, that is, it slightly increases the disorderer events. Coarse grained simulations50 have shown that even a small perturbation of the polypeptide free energy landscape, for example, a fraction of kcal/mol, dramatically influences the kinetics and the thermodynamics of the aggregation process. These simulation results could explain why a small perturbation produces the macroscopic decrease of amyloid aggregation observed in vitro in presence of inhibiting compounds. On the other hand, the slowing down or the inhibition of the ordered aggregation process, can multiply the alternative pathways for the aggregation,51 increasing the chance to create other toxic species. For this reason, blocking the formation of oligomeric soluble species at their very early stages (note that the smallest oligomer, the Aβ dimer, has been recently reported to be synaptotoxic52) should be a viable strategy, providing that the accumulation of the Aβ monomer would not trigger any significant toxic effect.53
One of the main goals of the present work is the understanding of the interactions between tricyclic compounds and ordered oligomers. Knowledge of such interactions may suggest modifications of AQ for improving its antiaggregation activity. New functional groups potentially able to establish additional strong interactions with Aβ could be added to the quinonic scaffold to improve the strength of binding. Among them, positively charged aminic groups, such as those present in anthracyclinic and xantronic antitumor drugs, might be selected to engage in strong ionic interactions and hydrogen bonds with polar groups of Aβ. Indeed, some anthracyclines have shown excellent antifibrillogenic activity.54 Moreover, pixantrone, an antitumor drug in phase III clinical trials for the treatment of relapsed or refractory indolent non-Hodgkin's lymphoma, showed activity and proved to selectively target soluble low oligomers of Aβ protein.55 Another suggestion, inspired by the present simulation results, would be to link AQ to one or both termini of a recently discovered dipeptide inhibitor of Aβ oligomerization.31 These experimental findings, together with the results obtained in the present study, provide useful insights for the design of new molecular entities targeting the early steps of formation of low oligomeric, toxic species.
Acknowledgements
The authors are grateful to F. Marchand and Dr. O. Nicolotti for interesting discussions and precious suggestions, Dr. R. Friedman for comments to the manuscript, and A. Widmer for providing the WitnotP program used for visual analysis of the trajectories. The simulations were performed on the Matterhorn cluster of the University of Zurich and the support of C. Bolliger and A. Godknecht is gratefully acknowledged.

