

Structural characterization of unphosphorylated STAT5a oligomerization equilibrium in solution by small-angle X-ray scattering
Abstract
Signal transducer and activator of transcription (STAT) proteins play a crucial role in the activation of gene transcription in response to extracellular stimuli. The regulation and activity of these proteins require a complex rearrangement of the domains. According to the established models, based on crystallographic data, STATs convert from a basal antiparallel inactive dimer into a parallel active one following phosphorylation. The simultaneous analysis of small-angle X-ray scattering data measured at different concentrations of unphosphorylated human STAT5a core domain unambiguously identifies the simultaneous presence of a monomer and a dimer. The dimer is the minor species but could be structurally characterized by SAXS in the presence of the monomer using appropriate computational tools and shown to correspond to the antiparallel assembly. The equilibrium is governed by a moderate dissociation constant of Kd ∼ 90 μM. Integration of these results with previous knowledge of the N-terminal domain structure and dissociation constants allows the modeling of the full-length protein. A complex network of intermolecular interactions of low or medium affinity is suggested. These contacts can be eventually formed or broken to trigger the dramatic modifications in the dimeric arrangement needed for STAT regulation and activity.
Introduction
Signal transducer and activator of transcription (STAT) proteins have the ability to transduce signals from the cell membrane to the nucleus where they activate gene transcription. The general features of STAT activity and regulation mechanism are known and have been extensively reviewed.1-4 STATs are activated in response to extracellular signals such as cytokines, growth factors, and hormones.5-7 Binding of these factors to cell surface receptors induces receptor autophosphorylation and recruitment of STATs by the receptor. STATs are then phosphorylated on a conserved C-terminal tyrosine mainly by JAK kinase family members,8 causing the subsequent translocation to the nucleus.9 This family of proteins is tightly regulated and it has been shown that constitutive activation of certain STATs or upregulation of phosphorylated STAT activity are associated with cancer.10, 11
Seven mammalian STAT genes have been identified both in human and mouse genomes, Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b, and Stat6. STAT genes encode for proteins of variable length, 750–850 residues, sharing a pairwise sequence identity of 20–50%.12 The domain arrangement is common to all STATs and encompasses five domains: the N-terminal domain, a coiled-coil domain, a DNA binding domain, a SH2 domain, and a transactivation domain. In addition to full-length (FL) proteins, several shorter proteins exist arising from alternative splicing or proteolytic processing.13 Three-dimensional structures of partial constructs of different STATs have been elucidated by X-ray crystallographic studies. The first structure reported was the N-terminal domain of STAT4, a helical domain that crystallized as a dimer.14 The structures of the phosphorylated (active) STAT1 and STAT3 core fragments, lacking the N-terminal and the C-terminal transactivation domains, were solved in the DNA-bound form as dimers.12, 15 Both constructs contain a very elongated and slightly bent four-helix bundle of antiparallel topology connected by short loops. Next to it, there is an eight-stranded β-barrel domain that is responsible for DNA binding. The β-barrel domain is connected to the SH2 domain through a small helical domain that is known as “connector” or linker domain.
The three-dimensional structures of unphosphorylated STAT1 and STAT5a individual molecules are very similar to the reported phosphorylated STATs. The RMSD of STAT5a with the phosphorylated STAT1 and STAT3 is only around 2.1 and 2.3 Å, respectively.16 However, the dimeric arrangements in the crystals of phosphorylated and unphosphorylated forms are completely different. Phosphorylated STAT1 and STAT3 dimers, crystallized in the presence of DNA, adopt a parallel arrangement driven by the intermolecular phosphotyrosine-SH2 interaction, and the simultaneous contact of both β-barrel domains with the double-stranded DNA. The dimer adopts a partially open shape where both coiled-coil domains extend out from the DNA binding region. Conversely, unphosphorylated STAT1 and STAT5a core domain dimers present an antiparallel architecture burying a large interface between monomers with complementary shapes. The interaction surface mainly includes hydrophilic residues from the DNA-binding and the coiled-coil domains.16 Unphosphorylated STAT1 including the N-terminal domain, presents a tetrametric architecture where the two antiparallel dimeric core domains interact via the N-terminal domain dimers.17 A very recent structure of the unphosphorylated STAT3 core domain seems to be in disagreement with the antiparallel arrangement found for other unphosphorylated STATs.18 STAT3 core domain crystallized forming a parallel assembly with the two SH2 domains interacting. However, in contrast to the parallel dimers found in phosphorylated STAT1 and STAT3, the DNA-binding domains are in the opposite sides of the dimer leading to a less open arrangement. Additionally, the structure of a phosphorylated STAT protein from Dictyostelium discoideum in the absence of DNA has also been reported.19 Despite the low homology with mammalian STATs, around 15%, and the noticeable shorter four-helix bundle domain, the overall structure is very similar to the mammalian STATs. The Dictyostyelium STAT dimer is stabilized by intermolecular phosphotyrosine-SH2 interactions but presents a fully extended shape that would require a 135° relative rotation to be superimposable to the DNA-bound open dimers of phosphorylated mammalian STATs. Given the low sequence homology, the question whether a similar dimeric arrangement would exists in mammalian STATs in the absence of DNA remains unclear.
Available crystallographic data combined with extensive mutagenesis studies led to a model for the rearrangement of STATs from the inactive to the active state and vice versa.20, 21 According to this model, transitions from parallel to antiparallel arrangements, implying dramatic modifications in the interaction regions, are mediated by the dimerization of the N-terminal domain. This concerted motion would most likely require a temporal breaking of the intermolecular core region interactions. Then, highly mobile dimeric species, only held together by the N-terminal dimerization domain, would allow the transition to the opposite assembly facilitated by the high flexibility of the linker connecting the N-terminal and the coiled-coil domains. However, a more detailed description at molecular level is necessary for a better understanding of these complex rearrangements.
The structure, regulation, and functioning of STATs basal state has attracted great interest. Recent findings indicate that in the basal state STAT3 can drive and coregulate transcription,22 and also perform other tasks in the cytoplasm such as microtubule-stabilizer.23 The roles of unphosphorylated STAT proteins in transcription and regulation of gene expression have been recently reviewed.24 These studies highlight an active role of unphosphorylated STATs that goes beyond the concept of a resting state waiting to be activated but pictures the basal STATs as species continuously shuttling between the cytoplasm and the nucleus.
Although it was initially reported that STATs were monomeric in their latent state,25 several studies indicate the presence of dimeric or higher molecular weight species in cell extracts or in vivo.26-30In vitro studies have demonstrated that several FL unphosphorylated STATs form stable dimers in solution.29, 31 The presence of heterodimers has been also demonstrated in vivo.27, 32, 33 Experiments with N-terminal domain point mutants indicate that the N-terminal domain is required for STAT4 dimerization,31 and point or deletion mutants unable to oligomerize were shown not to be phosphorylated in vivo.31, 34 Despite the X-ray structures, the importance of the core domain in the dimerization of unphosphorylated STATs in solution is unclear. Equilibrium sedimentation experiments suggested the coexistence of monomeric and dimeric forms for the core domain of STAT1.17 The dissociation constant of this core domain, Kd = 21.82 μM, is 30 times weaker than the one for the FL protein, Kd = 0.68 μM, but only three times weaker than the isolated N-terminal domain, Kd = 6.37 μM. Conversely, gel filtration chromatography and dynamic light scattering experiments suggest that STAT5a and STAT3 are mostly monomeric in solution.16, 18
Here we report a small-angle X-ray scattering (SAXS) study of the unphosphorylated core domain of STAT5a in solution. The integration of concentration dependent SAXS with appropriate computational tools allowed the structural and thermodynamic characterization of the equilibrium between monomeric and dimeric STAT5a in solution with a Kd ∼ 90 μM. The dimer present in solution corresponds to the parallel arrangement found in the crystals. Knowledge of the solution equilibrium of STAT5a gives new insights into the complex structure-based regulation of STAT proteins.
Results
Variation of STAT5a SAXS overall parameters with concentration
SAXS is a very well-suited technique to study biomolecules and their complexes in solution.35-37 SAXS curves measured for STAT5a at three different concentrations, 4.6, 2.3, and 1.15 mg/mL, are displayed in Figure 1, and Table I reports on the parameters that describe the overall properties derived from the measured curves. The apparent radius of gyration, derived either with the Guinier approximation,41 or from the distance distribution function using GNOM,39 increases with protein concentration beyond the experimental error. The Rg value estimated by GNOM at the lowest concentration, 38.1 Å, is in very good agreement with the theoretical Rg for the monomer calculated from the X-ray structure, 1Y1U, 38.06 Å. Conversely, the Rg calculated for the crystallographic dimer is 45.54 Å, larger than the Rg derived for the highest concentration sample, 43.7 Å. The maximum particle distance, Dmax obtained for the 1.15 mg/mL sample is 145 ± 5 Å, in good agreement with the theoretical value computed from the monomeric structure, 147.5 Å. For the highest concentration sample the value of Dmax obtained was 165 ± 5 Å which is also in good agreement with the dimeric structure, 162.7 Å. The analysis of the apparent R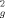 as a weighted average of the squared Rgs estimated from the crystallographic monomeric and dimeric forms suggests a dissociation constant which is on the μM range. More accurate estimates for the thermodynamic constant can be obtained with approaches that use the whole dataset and momentum transfer range.
as a weighted average of the squared Rgs estimated from the crystallographic monomeric and dimeric forms suggests a dissociation constant which is on the μM range. More accurate estimates for the thermodynamic constant can be obtained with approaches that use the whole dataset and momentum transfer range.

Scattering intensities as a function of the momentum transfer s = 4π sin(θ)/λ for STAT5a at different concentrations 4.6 mg/mL (A), 2.3 mg/mL (B), and 1.15 mg/mL (C). Fitted curves with the structures of the monomer (red solid lines) and dimer (green solid line) of the STAT5a (1Y1U) using CRYSOL are displayed. The curves are appropriately displaced along the axis for better visualization. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
| STAT5a Conc. (mg/mL) | Rg (Å)a | Dmax (Å)b | Rg (Å)b | MW (KDa)c | χ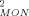 d d |
χ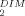 e e |
|---|---|---|---|---|---|---|
| 4.6 | 40.2 ± 0.1 | 165 ± 5 | 43.7 | 71 | 49.00 | 81.90 |
| 2.3 | 38.2 ± 0.2 | 155 ± 5 | 41.3 | 61 | 8.53 | 41.86 |
| 1.15 | 35.5 ± 0.5 | 145 ± 5 | 38.1 | 49 | 1.74 | 8.64 |
- a Rg derived with the Guinier approximation using PRIMUS.38
- b Parameter derived with GNOM.39
- c Apparent molecular weight obtained by comparison of the forward scattering, I(0), with that of a bovine serum albumin sample (3.7 mg/mL).
- d Fitting error of the scattering curve to the crystallographic monomer structure of STAT5a, 1Y1U using16 CRYSOL.40
- e Fitting error of the scattering curve to the crystallographic dimer structure of STAT5a, 1Y1U16 using CRYSOL.35
The forward scattering, I(0), derived from Guinier's approximation, provides an estimate of the apparent molecular weight, which is a useful parameter to explore the oligomerization state of proteins in solution. The analysis of the apparent molecular weight for the three concentrations, displayed in Table I, shows a systematic increase with the concentration of the sample, suggesting a concentration dependent equilibrium in which the major species is the monomer.
The scattering profiles were fitted to the crystallographic monomer 1Y1U16 using the program CRYSOL.40 Results are displayed in Figure 1 and χ2 values are given in Table I. The curve at 1.15 mg/mL is in relatively good agreement with the monomer structure, χ2 = 1.74. However, the agreement decreases at higher concentrations. Attempts to fit the data using only the structure of the crystallographic dimer were unsuccessful at any of the concentrations studied; see Figure 1 and Table I.
The variation of complex response patterns in a series of experiments related by the controlled variation of a single parameter can be analyzed using Principal Component Analysis (PCA). PCA has been applied to SAXS data in folding/unfolding, and protein self-association studies.42-45 PCA provides an unbiased estimation of the number of species contributing to the experimental data. The PCA of the three experimental SAXS profiles yields the three eigenvectors shown in Figure 2. The two eigenvectors with the largest amplitudes clearly display noticeable features, whereas the third one shows random values around I(s) = 0. These results suggest that the experimental profiles do not contain contributions of additional species and can be safely described as a combination of two scattering profiles. Nevertheless, at this level of precision the presence of a third species at very low concentration can not be excluded from the PCA. Similarly, the presence of multiple species with the same (or very similar) dimerization constants so that their relative populations do not change with concentration would appear as a single species. However, the similarity observed between a single dimer species giving a good fit to the experimental data and the crystallographic dimer (see later) strongly supports the claim of a dominant monomer–dimer equilibrium.

Eigenvectors resulting from the principal component analysis of the experimental scattering profiles. The third eigenvector (bottom panel) takes random values around I(s) = 0.0 indicating that the experimental data can be properly described as the contribution of only two species.
In summary, the variation of the overall parameters with concentration, and the PCA suggest the coexistence of two species in equilibrium, with monomeric STAT5a as the major species. This is in agreement with previous analytical ultracentrifugation studies showing a monomer–dimer equilibrium for this protein in solution.16 However, ultracentrifugation studies do not provide the structure of the dimer. In the next section, we show that this information can be extracted from SAXS data from the equilibrium mixture, even when the dimer is the minor species in solution.
Low-resolution structure of STAT5a dimer in a monomer–dimer equilibrium
The structural restrictions for the dimeric arrangement imposed by the experimental scattering profiles were explored using 5000 rigid body dimers which can be considered as a random distribution of possible assemblies. The ensemble was computed with the program FT-Dock.46 Although there are methodologies to obtain more physically meaningful and energetically refined ensembles, the aim at this stage was having a ‘complete’ survey of dimeric particle sizes and shapes. In fact, the pool of potential dimers covers a Rg range of 47.2 Å, from 39.4 Å for the most compact dimer to 86.6 Å for the most extended one. The scattering profiles at different concentrations were simultaneously fitted to a mixture of the crystallographic monomer with each one of the potential dimers in the ensemble. The dissociation constant, Kd, determines the relative population of both species at each concentration and is the only fitted parameter associated to each dimer. The quality of the fitting was assessed by the figure of merit, χ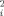 , see methods section for details of the optimization. Figure 3 displays the χ
, see methods section for details of the optimization. Figure 3 displays the χ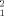 values associated to each of the 5,000 dimers, characterized by their Rg and the Kd value giving the best fit. Figure 3(A), which relates Rg and χ
values associated to each of the 5,000 dimers, characterized by their Rg and the Kd value giving the best fit. Figure 3(A), which relates Rg and χ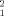 , shows that the best solutions reproducing the experimental SAXS data fall in a narrow range of Rg, 46.4 ± 1.8 Å for the best fifty solutions. It should be emphasized that only a small fraction of the putative dimers with Rg values in this range can explain the experimental SAXS profiles. This shows that the SAXS data contain additional structural information beyond Rg. Figure 3(B) shows the optimal Kd values for each of the 5000 putative dimers together with their χ
, shows that the best solutions reproducing the experimental SAXS data fall in a narrow range of Rg, 46.4 ± 1.8 Å for the best fifty solutions. It should be emphasized that only a small fraction of the putative dimers with Rg values in this range can explain the experimental SAXS profiles. This shows that the SAXS data contain additional structural information beyond Rg. Figure 3(B) shows the optimal Kd values for each of the 5000 putative dimers together with their χ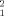 value. The best fits neatly define the optimal thermodynamic constant, and the top fifty solutions define a narrow range of dissociation constants, Kd = 86 ± 11 μM. As shown for the Rg values, a good fit cannot be obtained simply adjusting the concentration of the species present, using the correct Kd, but it requires choosing the correct structure for the dimer. The best fifty solutions, with χ
value. The best fits neatly define the optimal thermodynamic constant, and the top fifty solutions define a narrow range of dissociation constants, Kd = 86 ± 11 μM. As shown for the Rg values, a good fit cannot be obtained simply adjusting the concentration of the species present, using the correct Kd, but it requires choosing the correct structure for the dimer. The best fifty solutions, with χ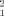 = 8.36 ± 0.79, are confined in a very narrow area of the Rg versus Kd plot displayed in Figure 3(C).
= 8.36 ± 0.79, are confined in a very narrow area of the Rg versus Kd plot displayed in Figure 3(C).

Structural restrictions in the dimer structure from experimental SAXS data. Each point represents the result of the optimization of the simultaneous fit of the scattering profiles at different concentrations using combinations of the crystallographic STAT5a monomer and one of 5000 different dimeric arrangements. For each dimeric arrangement, an optimal dissociation constant (Kd) and a figure of merit (χ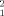 ) were obtained. (A) correlation of χ
) were obtained. (A) correlation of χ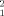 with Rg. (B) correlation of χ
with Rg. (B) correlation of χ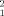 with Kd. (C) Combined Kd and Rg values of the best 50 solutions. The total plot area corresponds to the ranges of Kd and Rg obtained in the 5000 minimization, 39.41 Å < Rg < 86.57 Å, and 0.1 μM < Kd < 212 μM. (D) Most representative members of the structural clusters including the 50 structures with the best figures of merit χ
with Kd. (C) Combined Kd and Rg values of the best 50 solutions. The total plot area corresponds to the ranges of Kd and Rg obtained in the 5000 minimization, 39.41 Å < Rg < 86.57 Å, and 0.1 μM < Kd < 212 μM. (D) Most representative members of the structural clusters including the 50 structures with the best figures of merit χ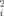 . The populations of the respective clusters are, from the left to right and from top to bottom, 20, 13, 8, 7, and 2.
. The populations of the respective clusters are, from the left to right and from top to bottom, 20, 13, 8, 7, and 2.
The 50 dimers that provide the best agreement with the SAXS data were clustered in five structurally related families, Figure 3(D). This analysis was done using the normalized spatial discrepancy (NSD) that monitors the mass distribution in the space, and that has been proven to be highly efficient to analyze SAXS-derived structural models,47, 48 see definition in methods section. The different clusters are not homogeneously populated, having from 20 to two members. The most populated cluster is highly similar to the antiparallel structure found for unphosphorylated STAT117 and STAT5a.16 The average NSD between the X-ray STAT5a dimer and the members of the cluster is 1.65. This is a remarkable result taking into account that no symmetry restraints were imposed in the docking calculation of putative dimers. Figure 4(A) shows that the STAT5a crystal structure perfectly reproduces the experimental data.

Point-by-point error of the simultaneous fitting of three SAXS curves to a monomer–dimer equilibrium including the crystallographic monomer and one of five different dimeric models of STAT5a built by homology from arrangements found in crystallographic studies of different STATs. (A) Antiparallel structure of STAT5a,16 (B) alternative dimer found in STAT5a crystallographic cell formed with the third monomer of the asymmetric unit, and probably resulting from crystallographic contacts.16 (C) Parallel arrangement of unphosphorylated STAT3, 3CWG.18 (D) Putative dimerization mode for STAT5a built by homology from DNA bound phosphorylated STAT3, 1BG1.12 (E) Putative dimerization mode derived from phosphorylated Dictyostelium STAT, 1UUR.19 The curves measured at 4.6, 2.3, and 1.15 mg/mL correspond to the top, middle and bottom panels, respectively.
Compatibility of the solution data with the crystallographic forms of the STATs
Five different monomer-dimer equilibrium models using dimers found in crystal structures of different STATs were specifically tested for their ability to describe the SAXS curves at different concentrations using the methodology described in the previous section. STAT dimers have been crystallized in different arrangements depending on the phosphorylation state and the presence of DNA. The five dimeric assemblies used are displayed in Figure 4. The asymmetric unit of STAT5a crystals contains three molecules that give rise to two different dimeric arrangements.16 The one reported as biologically relevant is equivalent to the previously found for STAT1. It is symmetric and has a large dimeric interface, see Figure 4(A). A second dimeric arrangement, probably resulting from crystallographic contacts, has a three-fold smaller interaction surface, see Figure 4(B). Three other potential dimeric arrangements for STAT5a were built by homology from the X-ray structures of other STATs. Figure 4(C) displays the protein dimer modeled from the parallel arrangement recently found for the unphosphorylated STAT3.18 Figure 4(D) shows the partially open parallel structure based on DNA-bound STAT3β,12 and Figure 4(E) shows the completely open dimer modeled from Dictyostelium STAT structure.19
The point-by-point error function, Figure 4, clearly demonstrates that the parallel arrangement found in STAT5a X-ray structure is able to simultaneously describe the different SAXS curves, χ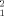 = 8.72, with no systematic departure from the horizontal Error = 0 line. The theoretical Rg for this structure is 46.21 Å, in agreement with the optimal value obtained using the systematic exploration of potential dimeric arrangements, see Figure 3(A). The best fitted value for the dissociation constant, Kd = 78 μM, is also in the optimal range found with the systematic exploration, 86 ± 11 μM.
= 8.72, with no systematic departure from the horizontal Error = 0 line. The theoretical Rg for this structure is 46.21 Å, in agreement with the optimal value obtained using the systematic exploration of potential dimeric arrangements, see Figure 3(A). The best fitted value for the dissociation constant, Kd = 78 μM, is also in the optimal range found with the systematic exploration, 86 ± 11 μM.
Interestingly, the alternative dimerization mode obtained in the X-ray structure of STAT5a has a similar Rg, 48.55 Å. Despite this similarity, the point-by-point error function shows important systematic deviations for the three SAXS curves. The larger χ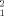 , 95.36, confirms that this crystallographic arrangement is not populated in solution. The same conclusion can be derived for the parallel arrangement modeled from unphosphorylated STAT3 that has a Rg = 45.19 Å. This exemplifies the sensitivity of SAXS data to the global shape of particles and not just the size. Likewise, the homology models representing dimeric arrangements found in phosphorylated STAT proteins fail to explain the STAT5a SAXS data in solution. This is not a surprising result taking into account that their theoretical Rg are 50.42 and 66.04 Å, respectively, larger than the optimum Rg.
, 95.36, confirms that this crystallographic arrangement is not populated in solution. The same conclusion can be derived for the parallel arrangement modeled from unphosphorylated STAT3 that has a Rg = 45.19 Å. This exemplifies the sensitivity of SAXS data to the global shape of particles and not just the size. Likewise, the homology models representing dimeric arrangements found in phosphorylated STAT proteins fail to explain the STAT5a SAXS data in solution. This is not a surprising result taking into account that their theoretical Rg are 50.42 and 66.04 Å, respectively, larger than the optimum Rg.
Discussion
STAT proteins play a crucial role in the activation of gene transcription in response to stimuli originating outside of the cell. The regulation and activity of STAT proteins require a complex rearrangement of the domains that, according to the established models, transform an antiparallel inactive dimer into a parallel active one.
Using different approaches we have addressed the study of human STAT5a core domain in solution. SAXS in combination with appropriate computational tools has been proven to be well-suited to study polydisperse samples such as those arising from oligomerization equilibrium in solution. Variation of the overall size descriptor parameters Rg, Dmax, and the apparent molecular weight with protein concentration clearly suggests the presence of an oligomerization equilibrium in solution. Previous knowledge based on in vitro and in vivo assays, as well as crystallographic results suggest that the equilibrium observed is between a monomer and a dimer. The absence of additional species in equilibrium was further substantiated using PCA.
The experimental SAXS curves could be quantitatively explained with a monomer-dimer model with a dissociation constant of 86 ± 11 μM. This value of Kd is comparable with the one obtained using analytical ultracentrifugation experiments for the core domain of STAT1, 21.8 ± 12 μM.17
The analysis of the SAXS curves at different concentrations also provides information about the structure of the dimer despite the low population of this species in solution. A large pool of 5000 different potential dimeric arrangements were tested and an optimal Rg for the dimer around 46 Å was found, in excellent agreement with the crystallographic structure, 46.21 Å. Although the low-resolution nature of SAXS data is not enough to define unambiguously the structure of the dimer, the most populated cluster of dimers obtained from the analysis is very similar to the STAT5a crystallographic structure. When an equivalent analysis is performed with alternative crystallographic dimeric structures obtained for different STAT proteins, the antiparallel arrangement found for unphosphorylated STAT5a and STAT1 was the only one able to describe the data measured in solution.
From a methodological point of view this study demonstrates the capability to derive structurally relevant information about a species representing less than 50% of the total protein in the most concentrated sample. Increasing the sample concentration to favor dimer formation, while desirable in principle, is limited by problems associated with interparticle interactions and nonspecific aggregation. A concentration of 270 mg/mL of STAT5 would be needed to have 90% dimer is solution. The approach presented here can be important for the structural characterization of low and moderate affinity biomolecular complexes.
The assessment of the antiparallel dimer as the relevant arrangement in the basal state as well as the strength of the interaction allows the modeling of the FL protein (FL-STAT5a). A large ensemble of 5000 conformations of the FL-STAT5a was built with Pre_Bunch49 assuming that the protein encompasses two rigid domains, the N-terminal and the core domain, tethered by a highly flexible linker. Figure 5 shows this ensemble built on the antiparallel architecture for the core domain. Only the Cα atoms from residue 78 are displayed representing the whole N-terminal domain. This residue is conserved amongst all STATs but STAT6 and plays a key role in the dimerization of the N-terminal domain, as shown by X-ray and mutational studies.17, 20, 31, 50 Figure 5 shows the broad region around the core domain sampled by the N-terminal domain, as a consequence of the flexibility of the 14-residue linker. In the antiparallel dimer, the space sampled by both N-terminal domains overlap and, therefore, dimerization of the N-terminal and core domains can happen simultaneously. However, the overlapping region is small implying that the simultaneous dimerization of both domains would result in an important entropic penalty that could destabilize the doubly-anchored dimer. The dynamic nature of STAT dimers, where the formation of one dimeric interface destabilizes the other, could be a key element for the dramatic rearrangement associated to activation and DNA binding.51 Thus, the core domain interface could be broken prior to phosphorylation,17, 31 without loosing the overall dimeric stoichiometry. In addition, this double connection could avoid generating mixed STAT dimers that might complicate signaling specificity. All STAT proteins, with the exception of STAT6, present connecting linkers of similar length suggesting that the presented model could be generalized to other STATs.

Three orthogonal views of a model ensemble of full-length STAT5a. The blue and red envelopes represent the STAT5a core domain monomers in the antiparallel dimer. Dots represent the position of the Cα atoms of residue L78 of the N-terminal domain in 5000 different models generated with Pre_Bunch using the N-terminal structure of STAT4 and a 14 residue flexible linker. Cyan and orange dots correspond to the L78 residue from the blue and red units, respectively. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
This model is in agreement with previous biophysical and biochemical studies. It could explain the analytical ultracentrifugation results obtained for STAT1.17 In this study Mao et al. determined the dissociation constants for the N-terminal domain, the core domain and the FL protein to be 6.4 ± 1.3, 21.8 ± 12, and 0.68 μM, respectively. In a scenario where both domains can dimerize independently, the expected Kd for the FL protein should be <1.0 nM. We hypothesize that the apparent destabilization of the FL-STAT1 dimers could originate from the entropic loss induced by the formation of the N-terminal dimer. The model is also in agreement with in vitro results by Mertens et al. that analyzed the phosphorylation and dephosphorylation kinetics of STAT1 constructs with engineered versions of the tether.21 Constructs with notably shorter linkers maintained the capability of being phosphorylated, but were unable to be dephosphorylated. Conversely, linkers of the same length, but with reversed or completely different amino acid sequences showed normal phosphorylation and dephosphorylation kinetics. These results suggest that it is the length of the linker and not the specific sequence that is important to promote the structural rearrangement necessary for dephosphorylation.
In summary, the structure and thermodynamics of the human STAT5a core domain in solution have been studied in solution by SAXS confirming the antiparallel structure of the dimer found in the crystal structure. When the flexibility of the linker domain is taken into consideration this structure suggests a structural hypothesis for the complex structural rearrangements necessary to adopt the active (phosphorylated) state from the basal (unphosphorylated) one. The inherent flexibility of STAT regulation process makes it a challenging one to study by high resolution methods. In that sense, we envision a relevant role of SAXS in unraveling the intricate regulation mechanism of STAT proteins.
Materials and Methods
STAT5a expression and purification
The sequence coding the fragment 129–712 of human STAT5a was amplified by PCR and the product cloned into the Nco1-Nde1 site of the expression vector pET14b (Novagen). The expression construct was transformed into the Escherichia coli strain BL21(DE3). Protein expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside, and after 12 h at 25°C the cells were harvested by centrifugation. The cell pellets were resuspended in buffer A (20 mM Tris, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 1 mM DTT, pH 8.0) and lysed by sonication. The lysate was clarified by centrifugation at 25,000g during 1 h. The supernatant was diluted with buffer B (20 mM Tris-HCl, pH 8.0, 1 mM DTT, 1 mM EDTA, 5% glycerol), applied to a Q-Sepharose FF column (Amersham) and STAT5a was eluted with a gradient from 0.05 to 1M NaCl. Protein containing fractions were concentrated, (NH4)2SO4 was added to a concentration 900 mM and the solution was passed over a 20 mL Phenyl-Sepharose FF column (Amersham) equilibrated with 900 mM (NH4)2SO4 in buffer C (20 mM Tris-HCl, pH 8.0, 1 mM DTT, 1 mM EDTA, 5% glycerol). After washing with equilibration buffer, the protein was eluted decreasing the salt concentration to zero. After elution, the protein fractions were combined, diluted with buffer C, concentrated to 10 mL, applied to a heparin affinity column (Amersham), and eluted using a gradient from 0.05 to 1M NaCl in buffer C. The protein obtained was loaded on a HiLoad Superdex 200 gel filtration column, which had been equilibrated with buffer D (20 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT). Fractions containing STAT5a were then concentrated prior to SAXS measurements. After purification, the sequence of purified protein was confirmed by tryptic digestion and peptide mass mapping by mass spectrometry. The N-terminus of purified STAT5a was checked by Edman sequencing: the protein was electrophoresed on a 6% SDS-PAGE gel and electrotransferred onto a PVDF membrane.
SAXS measurements and overall parameter determination
Small-angle X-ray scattering data were collected at X33 beamline at the European Molecular Biology Laboratory (EMBL) in the storage ring DORIS III of the Deutsches Elektronen Synchrotron (Hamburg).52 Scattering curves of human STAT5a were measured at 20°C at protein concentrations of 4.6, 2.3, and 1.15 mg/mL, and an exposure time of 3 min. The scattering profiles covered a range of momentum transfer of 0.02 < s < 0.5 Å−1. Radiation damage was monitored by repetitive three-minute exposures of protein solutions, and no significant changes were observed. Buffer scattering profiles were measured before and after the samples, they were averaged, and used for the subtraction from the protein scattering profiles. All data manipulations were done using standard procedures with the software PRIMUS.38
The forward scattering, I(0), and the radius of gyration, Rg, were evaluated using the Guinier approximation,41 assuming that at very small angles (s < 1.3/Rg), the intensity can be well-represented as I(s) = I(0) exp(−(sRg)2/3). The maximum particle dimensions, Dmax, were computed from the entire curves with the program GNOM.39
The apparent molecular weights of the proteins were estimated from their forward scattering, I(0), by comparison to the one obtained for a Bovine Serum Albumin sample of 3.7 mg/mL.
Principal component analysis (PCA)
SAXS data of complex mixtures are concentration weighted linear combinations of the scattering curves arising from the different species present. Principal component analysis (PCA) of a series of experiments has been used to determine the minimal number of species that are able to describe the measured data. With only three scattering curves, a good PCA was supposed to be achieved by only using the best-defined region of the scattering curves, thus a momentum transfer range of 0.025 < s < 0.193 Å was used. The program MATLAB was used for the PCA calculation.
Generation and structural analysis of potential dimeric arrangements
The ensemble of 5000 dimers used to explore the arrangements compatible with the SAXS data was obtained with the program FT-Dock46 using the monomer of STAT5a 1Y1U16 as a template. This program uses a Fourier transform strategy to speed up the systematic screening of all possible interaction surfaces. A shape-complementarity term is included in the algorithm to score the resulting assemblies.
 ()
()Simultaneous fitting of the SAXS curves to a monomer–dimer equilibrium model
 term,
term,
 ()
() ()
() ()
()Modeling the FL STAT5a
An ensemble of 5000 conformers of the FL STAT5a monomer were built with Pre_Bunch49 using the STAT4 N-terminal (1BGF) and the STAT5a core (1Y1U) domains as rigid entities connected by a 14-residue long flexible linker. Each one of the members of the ensemble were translated and rotated onto both core domains of the antiparallel dimeric arrangement of STAT5a using the SHELXPRO software.53
Acknowledgements
Authors are in debt with Montse Soler-López (IRB) for stimulating discussions and with Isabel Usón (Institut de Biologia Molecular de Barcelona) for help in SHELXPRO usage. P.B. holds a Ramón y Cajal contract partially financed by the Spanish Ministry of Education. J.B. is a recipient of a predoctoral fellowship from the Spanish Ministerio de Educación y Ciencia. The authors acknowledge the support of the European Community—Research infrastructure Action under the FP6 “Structuring the European research area program contract number RII/2004/5060008” to the EMBL-Hamburg outstation that covered the travel and accommodation expenses at the EMBL-Hamburg.

