

Intrinsic local destabilization of the C-terminus predisposes integrin α1 I domain to a conformational switch induced by collagen binding
Integrin–collagen interactions play a critical role in cell adhesion processes. To gain insight into the mechanisms underlying collagen-induced conformational switches, we have undertaken a comparative NMR study between the human integrin α1 I and a gain-of-function E317A mutant. Our results, supported by thermodynamic measurements, suggest that intrinsically destabilized regions facilitate conformational rearrangements of integrin I domain. This study highlights the importance of exploring slow dynamics to delineate allosteric and binding events.
Abstract
Integrin–collagen interactions play a critical role in a myriad of cellular functions that include immune response, and cell development and differentiation, yet their mechanism of binding is poorly understood. There is increasing evidence that conformational flexibility assumes a central role in the molecular mechanisms of protein–protein interactions and here we employ NMR hydrogen–deuterium exchange (HDX) experiments to explore the impact of slower timescale dynamic events. To gain insight into the mechanisms underlying collagen-induced conformational switches, we have undertaken a comparative study between the wild type integrin α1 I and a gain-of-function E317A mutant. NMR HDX results suggest a relationship between regions exhibiting a reduced local stability in the unbound I domain and those that undergo significant conformational changes upon binding. Specifically, the αC and α7 helices within the C-terminus are at the center of such major perturbations and present reduced local stabilities in the unbound state relative to other structural elements. Complementary isothermal titration calorimetry experiments have been performed to derive complete thermodynamic binding profiles for association of the collagen-like triple-helical peptide with wild type α1 I and E317A mutant. The differential energetics observed for E317A are consistent with the HDX experiments and support a model in which intrinsically destabilized regions predispose conformational rearrangement in the integrin I domain. This study highlights the importance of exploring different timescales to delineate allosteric and binding events.
Abbreviations
-
- α1 I
-
- alpha1 I domain
-
- NMR
-
- nuclear magnetic resonance
-
- HDX
-
- hydrogen–deuterium exchange
-
- HSCQ
-
- heteronuclear single-quantum correlation
-
- MIDAS
-
- metal ion-dependent adhesion site
-
- THP
-
- collagen-like triple helical peptide
-
- ITC
-
- isothermal titration calorimetry
-
- CD
-
- circular dichroism.
Introduction
Collagen interactions with α1β1 integrin receptors play a key role in numerous cellular processes, spanning cell development to differentiation and hemostasis to immune responses.1-3 Integrin α1β1 is widely expressed in mesenchyme cells, the immune system, and a minority of epithelial tissues.3 Functionally, α1 is one of four collagen binding I domains containing β1 partners that includes α2, α10, and α11. Upon binding to a collagen peptide, the I domain of integrin α1β1 (α1 I) undergoes conformational changes4-7 similar to those observed in the α2 I domain,4-7 in which coordination of a collagen glutamate to the metal ion-dependent adhesion site (MIDAS) at the top of α1 I induces a shift from the “closed” (unliganded) to “open” (liganded) conformation. This switch induces allosteric changes in the C-terminus of the Rossmann-fold structure, with helix αC unfolding and helix α7 displacing 12 Å downwards. The resultant movement permits coordination to a second allosteric site within helix α7 to the β-subunit and propagates these structural changes throughout the entire integrin macromolecule.1
Despite the critical functional importance of collagen-integrin interactions in vivo, the mechanism of integrin binding to collagen requires elaboration at the structural, dynamic, and thermodynamic levels. In an attempt to gain mechanistic insights into the I domain activation process induced by collagen, several “gain-of-function” mutants that result in enhanced binding to collagen have been studied.8-12 Specifically, the α1 I mutant E317A has been characterized by high resolution X-ray crystallography13 and numerous binding assays report an increase of binding to collagen.11, 13 The residue E317 is located at the top of helix α7 and is linked to helix αC (R287) via a salt bridge, a stabilizing feature of the closed unliganded form. The E317A mutation eliminates this salt bridge and adopts a transition state between the α1 I closed-unbound form and the open-bound form. Indeed, an X-ray structure of the E317A mutant has revealed a novel conformation in which helix α7 is in the closed upward position, helix αC is unstructured (as observed in the open-bound form), and the metal ion is reported as adopting a unique penta-coordination.13 Although the gain in function is primarily ascribed to structural changes that are implicated in reducing the ligand binding barrier relative to wild type protein, dynamics must be considered when describing the mechanism by which a gain of activity occurs.
There is increasing evidence that conformational flexibility assumes a central role in the molecular mechanisms of protein-protein interactions.14-17 NMR is well suited for studying the conformation and dynamics of proteins and can provide information over a broad spectrum of timescales ranging from picoseconds to seconds and hours.14, 15 Conformational fluctuations on the micro to millisecond timescales have been shown to be critical determinants of biological processes in protein recognition and allosteric events.14-18 In this study, we explore the role of slower timescale dynamic events that occur in the unbound free protein to unravel specific mechanisms that precede collagen binding. Hydrogen–deuterium exchange (HDX) kinetics measured via NMR affords the advantage of characterizing slow conformational fluctuations in denaturant free environments and thereby provides site-specific information on local stability in the native state.19-23
In the current investigation, NMR HDX experiments demonstrate the importance of slow motions in terms of predisposing integrin to conformational changes upon binding. Our NMR HDX results suggest a relationship between regions exhibiting a reduced local stability in the unbound I domain and those that undergo significant conformational changes upon binding. The αC and α7 helices of α1 I are at the center of such major perturbations and have reduced local stabilities in the unbound state, relative to other structural elements. Significantly, a combined energetic and structural characterization suggests that E317A activation and enhancement of collagen binding affinity are primarily dynamic in origin. The latter includes intermediary timescale motions in helix αC and MIDAS, as well as propagation of slow conformational fluctuations to additional structural elements within the C-terminus. Our findings underscore the relevance of slow conformational dynamics, intrinsic to the free α1 I, and the concomitant reduction of local stability within regions of a conformational switch. The latter assumes a critical role in allosteric regulation presumably by decreasing the overall energetic penalty associated with ligand-binding interactions.
Results
HDX NMR experiments reveal conformational fluctuations in the I domain of human α1β1 integrin
HDX measurements of α1 I integrin provide detailed information regarding conformational fluctuations based on the amide NH protection factors (Pf).19, 23 Well-dispersed resonances in the [1H-15N]-TROSY spectrum (Supporting Information Figs. S1A and S4A) are characteristic of a well-folded protein.6 HDX measurements for 186 non-overlapping residues in α1 I reveal 32% fast-exchanging amide protons with low Pf, and 23% slowly exchanging residues with high Pf [Fig. 1(A)]. The remaining 45% exhibit a time-dependent decrease in peak intensity and their observed exchange rate constants (kobs), Pf, and free exchange energies (ΔGHX) are summarized in Supporting Information Table SI.

Hydrogen–deuterium exchange of α1 I integrin. The logarithmic value of protection factors (Pf) obtained from the HDX exchange rates, are mapped in the representation of (A) wild type α1 I (PDB #1pt6) in the closed-unbound form. The residues that do not exchange with solvent for over two months exhibit high Pf (Log Pf > 7) and are colored in blue, whereas residues that are highly dynamic (i.e., exchange with solvent so fast that these disappear from the NMR spectra after 20 min) exhibit reduced Pf (Log Pf < 4) and are colored in red (refer to color bar). (B) Top view highlighting the flexibility of residues located at the top of collagen binding site in α1 I. Note that the blue color is removed for ease of visualization. Unassigned or overlapped peaks are colored in gray and the metal is represented as a sphere.
There are four major observations that can be deduced from the HDX kinetics data of α1 I [Fig. 1(A), Supporting Information Table SI]. First, α1 I contains a highly protected β-sheet core with amide protons that exhibit Pf values of 106−107 [Log Pf = 6–7 in Fig. 1(A)] comparable in magnitude to those observed for typical 10–20 kDa proteins.24, 25 Second, residues located in the upper face possess lower Pf values with faster HDX exchange rates than residues in the lower face [Fig. 1(B)]. Most of these fast-exchanging residues comprise unstructured loops and α-helical structures with the exception of helix α3. Third, highly protected loops of α3-βC and α6-βF are located in the bottom face (Pf of 105−106 compared to 102−104 in other loops), consistent with the existence of a hydrophobic intramolecular pocket observed in other I domains.26-29 Fourth, fast HDX rates are observed in helices αC and α7. Indeed, these residues present remarkably low Pf values for a helical conformation (104−105). In fact, only G283 in helix αC (G283SYNR287) exhibit measurable Pf values on the order of 105. These two helices are known to undergo a major structural rearrangement upon complexation [Fig. 2(A)].7, 30 Thus, the data suggest a relationship between fast HDX rates in the unbound α1 I and residues that undergo significant conformational change upon complexation. HDX can be interpreted in terms of the free exchange energies or local stability (ΔGHX) by assuming an EX2 limit,31 whereby the conformational equilibrium between unfolded and folded states is much faster than the intrinsic exchange rate. HDX will be discussed within the context of local stability (ΔGHX) in subsequent sections of this manuscript.

Relationship between conformational change upon complexation for α1 I residues, local stability, and solvent accessibility (SASA). (A) The X-ray α1 I closed-unbound structure (gray, PDB #1pt6) is overlaid with the first conformer of the GLOGEN bound open α1 I NMR structure (blue, PDB #2m32). The stick representation of E317 (orange) and R287 (cyan) residues highlight their distinct positions before and after collagen binding. (B) Correlation between α1 I free energy of exchange (ΔGHX) obtained from the protection factors, solvent accessible surface area (SASA), and distance between the alpha-carbon (ΔrCαopen/closed form) in the closed and open structures of α1 I taken from the X-ray6 and NMR7 structures. The C-terminal residues are colored in green except for residues assigned to helix-7 (orange) and C-helix (cyan). Drop lines highlight residues that have ΔrCα > 5 Å, ΔGHX < 6 kcal/mol and SASA < 0.5. Helices that have unusually low local stabilities (i.e., αC and α7 helices in the closed form) undergo large structural changes upon binding to GLOGEN.
Low local stabilities derived from HDX experiments can arise from increased solvent accessibility. We have therefore investigated the relationship between local stabilities derived from HDX experiments, solvent accessible surface areas (SASA), and conformational changes in the α1 I structure (ΔrCα) induced by collagen binding [Fig. 2(B)]. High ΔrCα values correspond to large conformational differences between the unbound-closed and bound-open forms of α1 I. In the overlay of both α1 I forms (Fig. 2), helix α7 translocates Å downward upon ligand binding and helix αC becomes an unstructured loop.7 Solvent accessible surface areas (SASA) derived from analysis of the three-dimensional structures measure solvent accessibility to an amide within the protein. Residues characterized by large SASA values are highly accessible to solvent and usually exhibit low Pf values (e.g., loops), while residues with low SASA values are buried from solvent. The most striking feature illustrated in this figure is the presence of residues (highlighted by drop lines) with low SASA that present low Pf values represented by reduced local stabilities (ΔGHX), all of which correspond to residues with the highest displacement (high ΔrCα). These residues are confined to the C-terminus of α1 I [Fig. 2(B)] and are primarily located in helices αC and α7.
Activated E317A α1 I mutant is more dynamic than its wild type counterpart
Our NMR data, using an E317A/α1 I sequence construct that is similar to the X-ray study, reveals 90% of the expected resonances (independent of the experimental conditions). The majority of missing resonances correspond to residues located in the L1 and L3 loops of MIDAS and helix αC and are attributed to conformational exchange dynamics on the micro-millisecond timescale (Supporting Information Figs. S1B and S1C). For the resonances that are observed, 13C chemical shift analysis32 suggests that E317A/α1 I retains a secondary structure in solution which is identical to the crystal structure13 (Supporting Information Fig. S1B) with the exception of helix αC and the MIDAS loops for which the NMR resonances are missing. Chemical shift difference spectra between E317A and the wild type protein support the notion that helix α7 is positioned upward as it is in the closed state (Supporting Information Fig. S1C). These results do not agree with previous NMR studies7, 33 that reported α7 in a downward position as in the open-bound form. This discrepancy may arise from the absence of reported peak assignments or from a different protein construct used in previous NMR studies relative to the crystal structure13 and our present NMR studies. The Scanlon group uses a construct exhibiting a truncation of the last three residues in helix α7 with the sequence terminating at I331.7
E317A/α1 I HDX experiments indicate that 31 and 20% of the residues are in fast and slow exchange, respectively and explicit kinetic parameters have been determined for 34% of these residues (Supporting Information Table SI). Comparison of the HDX parameters for E317A/α1 I and α1 I [Figs. 1(A) and 3(A)] indicate similar tendencies overall to those observed for α1 I; however, there are local regions in E317A that exhibit a decrease in Pf values. A more quantitative comparison of ΔGHX reveals that the HDX of the beta-sheet core residues in α1 I and E317A do not change appreciably, yet regions important for conformational rearrangement from the closed to open form exhibit both a lower average and narrower distribution of ΔGHX [Fig. 3(B)]. This is particularly significant in helix α6, strand βF, and helix α7, with the activating mutant in helix α7 exhibiting a lower and narrower distribution of ΔGHX. Indeed, the C-terminus secondary-structure elements present an overall destabilization of 5.6 kcal·mol−1 relative to wild type α1 I. The βF-strand ΔGHX median value is 4.0 kcal·mol−1 less stable in the mutant, while helices α6 and α7 are destabilized by 0.7 and 0.9 kcal·mol−1, respectively.

Hydrogen–deuterium exchange of α1 I activating mutant, E317A. (A) The logarithmic value of protection factors (Log Pf, refer to color bar) is mapped onto the crystal structure of the activated E317A/α1 I (PDB #4a0q) mutant. Unassigned or overlapped peaks are colored in gray, and the metal is represented as a sphere. A close-up of E317 and R287 residues comprising the salt-bridge in the wild type (right panel) appears on the left for E317A. (B) Boxplot diagram representing the dispersion of local free energy of exchange for amide protons (ΔGHX) of α1 I wild type (gray) and E317A (hatch white). This box represents the statistical distribution between 25% and 75% of the full sequence and the C-terminus with helices α6, α7, and βF strand plotted separately. The mean value is depicted by a ball and the whiskers represent maximum/minimum values of all data.
Energetics of wild type α1 I and E317A interactions with a collagen model peptide
Full-length collagen sequences contain multiple binding sites exhibiting variable integrin affinities9, 34, 35 that effectively precludes an accurate energetic characterization. Synthetic collagen-like triple-helical peptides (THP) represent a suitable model for biophysical5, 7, 30, 36-39 and functional studies9, 34, 35 of collagen. In an effort to elucidate the forces that promote α1 I binding to collagen and the origins of E317A enhanced activity observed in adhesion assays toward collagens I and IV (Supporting Information Fig. S2), we have employed ITC to derive complete thermodynamic binding profiles for association of Ac-(GPO)4GLOGEN(GPO)4GY-NH2 (GLOGEN) THP with wild type α1 I and the E317A mutant (Supporting Information Fig. S3). We specifically selected this high affinity GLOGEN binding sequence35 to provide structural, dynamic, and thermodynamic insight on the α1 I-collagen interaction.
Association of both I-domains with THP is characterized by formation of a 1:1 stoichiometric complex with favorable enthalpic and entropic contributions (Table 1). The Gibbs free energy of - 5.98 kcal·mol−1 determined for the α1 I-GLOGEN interaction yields a dissociation constant of ∼ 20 µM that corroborates solid phase binding studies.35 While wild type interactions are predominantly entropy-driven, peptide association with the mutant is characterized by nearly identical enthalpic and entropic components. The E317A activation of α1 I is accompanied by an approximately threefold enhancement in binding affinity, which is the result of a more favorable enthalpy (i.e., ΔΔH = −1.88 kcal·mol−1) and a concomitant loss in entropy (i.e., ΔTΔS = −1.32 kcal·mol−1). This characteristic enthalpy–entropy compensation yields a small net gain in the overall Gibbs binding free energy (ΔΔG) of −0.56 kcal·mol−1. Direct HDX characterization by NMR of the wild type α1 I-GLOGEN complex is not possible as the 20 µM dissociation constant determined for α1 I-GLOGEN interaction positions the interconversion between bound–unbound forms in an intermediate timescale, which results in resonance broadening within the NMR spectra of the complex (refer to Supporting Information Fig. S4). Nevertheless, the regions of α1 I most broadened by the addition of GLOGEN include the binding interface (particularly MIDAS, Supporting Information Fig. S4C,D) and the regions of structural rearrangement (primarily the C-terminus, Supporting Information Fig. S4C,E).
| Integrin | Kd (µM) | Ka 105 (M−1) | ΔG (kcal·mol−1) | ΔH (kcal·mol−1) | TΔS (kcal·mol−1) |
|---|---|---|---|---|---|
| Wild type α1 I | 19.9 ± 0.50 | 0.50 ± 0.01 | - 5.98 ± 0.15 | - 1.32 ± 0.02 | + 4.66 ± 0.10 |
| E317A/α1 I | 7.25 ± 0.13 | 1.38 ± 0.02 | - 6.54 ± 0.02 | - 3.20 ± 0.02 | + 3.34 ± 0.05 |
Discussion
The mechanisms that regulate α1 I ligand affinity of integrin and its intrinsic ability to undergo structural rearrangements upon binding to the rigid rod-shaped collagen remain unclear. Conformational fluctuations have been proposed as a driving force for several binding and allosteric events.14-17 The majority of these studies have focused on dynamic fluctuations occurring on the micro- to millisecond timescale14, 18, 40 in contrast to the impact of slower timescales on conformational changes. Much of what is known about allosteric activation of integrins caused by collagen binding has been derived from structural studies of both α1 and α2 I domains.5-7, 13, 41 These studies have captured static conformational “snapshots” of the collagen binding α1 I in three different conformations: a closed-unbound structure determined by X-ray,6, 41 a transitional conformation adopted by the gain-of-function E317A/α1 I mutant determined by X-ray,13 and an open-bound form determined by solution NMR with a complexed GLOGEN THP.7 The first glimpse of α1 I slow dynamics originated from a HDX mass spectroscopy study on a α1 I rat-human chimera and provided important insights on the impact of different ligands on integrin activation.42 To assess the role of intrinsic conformational fluctuations on the structural switch, our studies focus on elucidating the dynamics of unbound human α1 I integrin and its activating E317A mutant at the atomic level by HDX NMR spectroscopy in combination with ITC to evaluate the binding energetics. The resultant data suggest that slow timescale motions may be an integral determinant of αI-domain propensity to undergo significant structural changes upon collagen binding.
Intrinsic local destabilization of α1 I in the unbound-closed form facilitates a conformational switch to the open form
The difference in local stability as seen by HDX experiments between the upper and bottom face of the α1 I is quite significant, with the region that binds collagen,7, 42 at the top of α1 I, undergoing much faster exchange rates [Fig. 1(B)]. We propose that this increase in plasticity of the collagen binding interface might be required to allow the globular I domain to incorporate the rigid rod-like shape of the collagen structure. These slow timescale motions probed by HDX experiments are important for collagen recognition mechanisms in matrix metalloproteinases37 and may thereby assist in optimizing α1 I-collagen interactions.
The unusually low local stability of αC and α7 in wild type α1 I reveals that these helices undergo a local breathing or accordion-like motion. Notably, these helices are at the center of major structural rearrangements in the conformational switch of α1 I. In the complex, helix αC becomes unfolded and helix α7 exhibits a major displacement of 12 Å. The highly dynamic character of helix α7 is consistent with NMR and molecular dynamics studies of other I domain integrins.43-46 The data suggest a correlation between residues prone for allosteric movement and reduced local stability despite low solvent accessibility. We hypothesize that local destabilization of structural elements in the unbound I domain facilitates the conformational rearrangement induced by collagen.
Enhanced dynamics of unbound E317A/α1 I contributes to gain of functionality
To test our hypothesis we used an activated mutant of α1 I, E317A, that has been characterized by X-ray crystallography13 and affinity assays.11, 13 It provides mechanistic insight into the α1 I activation process due to its increase of affinity toward collagen and its transitional conformation.13 Here, we hypothesize that the increased binding affinity is also the result of higher local destabilization of the regions of conformational rearrangement.
NMR HDX measurements described in this study reveal that the E317A dynamics are more complex than wild type α1 I, particularly in the region of αC and MIDAS loops. Helix αC is a unique structural element of collagen-binding integrins that is proposed to be a determinant of selectivity for collagen5, 47, 48 by functioning via steric hindrance.11 In wild type α1 I, NMR resonances are observed for helix αC, whereas the latter are not detected in E317A. Signal loss provides valuable dynamic information, as peak broadening observed within the activating mutant indicates that helix αC and MIDAS loops are in conformational exchange.14, 18 In addition, the absence of peaks for helix αC, rather than the existence of sharp resonances expected for unfolded regions, implies that these residues are in intermediate exchange on the NMR timescale rather than fully unfolded as observed in the X-ray structure (Fig. 2). The E317A chemical shifts surrounding the MIDAS loops (refer to Supporting Information Fig. S1C) support a chemical environment that is distinct from wild type α1 I. Thus, we propose that the conformation of helix αC and MIDAS loops are in conformational exchange in solution, and that helix αC may be interconverting between folded-unfolded conformations on a fast micro-second to millisecond timescale, reducing the steric hindrance to collagen.
On the slower timescale motions, our HDX results on the activating mutant E317A/α1 I reveal a propagation of the trends already observed in wild type α1 I, and reflect an even lower and narrower range of local stabilities for the C-terminus residues involved in allosteric movement. The slow conformational dynamics are propagated from helix α7 to helix α6 and strand βF in the gain-of-function mutant, disturbing to a higher degree (than in wild type α1 I) the contacts within the hydrophobic intramolecular pocket.26-29 Indeed, destabilization of the hydrophobic core has been linked with an increase of binding toward collagen.28, 49 Moreover, deletion of three residues within helix α7 enhances the binding of α1 I to GLOGEN and allows visualization of the complex by NMR (refer to Supporting Information Fig. S4C).7 Higher destabilization of the E317A C-terminus coupled with the increase in binding affinity supports the hypothesis that these slow transient fluctuations observed in the unbound E317A/α1 I contribute to the gain-of-function by facilitating conformational rearrangement.
Thermodynamic basis of integrin-collagen interactions
Further corroboration of the forces driving association of α1 I with the THP may be deduced via comparisons of the respective thermodynamic profiles for both wild type α1 I and E317A mutant (refer to Table 1). The E317A mutant exhibits approximately a three-fold increase in affinity relative to wild type α1 I. The net enthalpic improvement of E317A association (i.e., ΔΔH = - 1.9 kcal·mol−1) compared to its wild type counterpart presumably results from abrogation of an unfavorable enthalpy that arises from local unfolding of the αC helix, and salt-bridge disruption that occurs upon collagen binding to the wild type. We suggest that hydrogen bonds are more transient in E317A due to absence of the salt bridge, faster dynamics of the αC helix and MIDAS loops, and the more destabilized C-terminus. Collectively, our findings on the E317A mutant suggest that the unbound form populates an ensemble of states that are conformationally more suited to ligand association, thereby reducing the overall energy (enthalpy) penalty imposed by major structural changes that must occur in the wild type.
Conclusions
Elucidating the forces that drive allostery is critical to understanding the complex transformations of biomolecules. The unique heterogeneous shape of a rod-like extracellular matrix protein associated with a globular cellular receptor prompts us to explore new timescales. HDX experiments on the wild type and activated α subunit I domains of integrin suggest that collagen binding and the induced conformational change are facilitated by destabilization of the C-terminus secondary-structure elements and residues comprising the integrin–collagen interface. Nature designed this domain with regions prone for conformational rearrangement given their inherent dynamics and intrinsic destabilization. Collectively, our characterization of slow dynamics in the integrin α1 I domain and the underlying binding energetics provides an instructive example on the relationship between local destabilization and propensity for allosteric structural changes. In an era where NMR microsecond timescale motions are critical for elucidating allosteric mechanisms, this study highlights the importance of exploring different timescales as part of a comprehensive experimental strategy to delineate allosteric and binding events.
Materials and Methods
I domains expression and purification
The recombinant α1 I from human integrin α1β1 used for these studies corresponds to residues T141-E335. Protein purification was conducted as previously described,12 but cells were grown in M9 media supplemented with 15NH4Cl2, and [13C6]-D-glucose and deuterated water was used when required to obtain isotopically 2H, 13C, 15N labeled proteins. Protein concentration was determined via measurement of the absorption at 280 nm employing the respective molar extinction coefficients.
NMR spectroscopy
Spectra were acquired on a 700-MHz Bruker spectrometer equipped with a cryoprobe. For experimental details refer to Supporting Information. Triple resonance experiments allowed assignments of 98 and 90% of the complete backbone resonances (195 residues) for wild type α1 I and E317A/α1 I, respectively, and agree with previous NMR studies of wild type α1 I.50 Chemical shifts of all assigned resonances of E317A/α1 I were deposited in the BMRB under accession number 26822. TALOS+51 was used to estimate the secondary structure in solution based on the 13C resonances. The analysis of 13Cα, 13Cβ, 13CO resonances32 reveals a comparable α1 I and E317A/α1 I secondary structure as in the crystal structures.6, 13, 41, 52 The chemical shift perturbation (Δδ) of the backbone amides caused by mutation of E317A/α1 I relative to wild type α1 I was calculated by the equation:
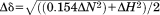 where ΔN and ΔH correspond the chemical shifts difference between the E317A's and the wild type's α1 I amide nitrogens and protons, respectively.53 All spectra were processed using nmr-Pipe54 and Sparky.55
where ΔN and ΔH correspond the chemical shifts difference between the E317A's and the wild type's α1 I amide nitrogens and protons, respectively.53 All spectra were processed using nmr-Pipe54 and Sparky.55
Hydrogen–deuterium exchange (HDX)
The amide exchange experiments were performed at 20ºC for the 15N-α1 I sample with a pD of 7.10 and at 25ºC for 15N-E317A/α1 I with a pD of 7.4. Samples were lyophilized in 10 mM PIPES buffer containing 140 mM NaCl, 5 mM or 25 mM MgCl2 and 1 mM DSS, with concentrations spanning the range 0.3-0.5 mM. A series of 1H-15N HSQC spectra of the D2O sample were acquired every 10 min for 24 hours, followed by several spectra of 1 hour duration up to 2 months for α1 I and 1 month for E317A/α1 I. Considering the time required to setup and acquire the NMR spectra, the first time points for α1 I and E317A/α1 I following resuspension in D2O were 15, and 20 min, respectively. HDX reaction of the amide proton is generally described by a two-step model between the folded (NHclosed) and unfolded (NHopen) states versus the exchanged (NHexchanged) state.31 The protection factor (Pf) of each amide proton was determined by a ratio of individual intrinsic rate constant (kint) for the intrinsic chemical HDX reaction of the freely exposed amide group and observed rate constant of exchange (kobs). The amide proton decays were monitored by plotting the peak intensities against the incubation times in order to obtain the kobs.56 Experimental uncertainties for kobs were obtained from fitting errors. For residues with Pf too low or too high to be quantified under our experimental conditions, the kobs values were estimated to be faster than kobs= (-Ln (0.05)/tmin) s−1 for the lower limit residues, where tmin is the first time point of each experiment considering that more than 95% of the signal intensity change has occurred. The upper limit residues were estimated to be slower than kobs= (-Ln (0.95)/tmax) s−1, where tmax is the time point of the last spectrum acquired. Thus, HDX kinetics were estimated to be faster than 3.0 × 10−3 s−1, and 2.3 × 10−3 s−1 for α1 I, and E317A/α1 I, respectively. The exchange in these residues is too fast for observation in the first NMR spectrum and too slow within the time frame of the experiments, thereby retaining their initial peak intensity. In contrast, the kobs value for residues that do not exchange after the last experimental point were estimated to be slower than 1 × 10−8 s−1 and 2.1 × 10−7 s−1 for α1 I and E317A/α1 I, respectively. The kint values were calculated from the amino acid sequence utilizing the methods of Bai et al57 and Connelly et al58 and the program SPHERE.59, 60 For α1 I, 5% of the residues were excluded from kinetic analysis due to unreliable data caused by severe resonance overlap. The exchange free energy of the amide protons was calculated from the equation ΔGHX= RT Ln (Pf), where R is the gas constant and T is the absolute temperature at which exchange was monitored. Under extreme conditions where the exchange rate is much faster than the refolding rate, ΔGHX estimated on the basis of EX2 limit would be larger than the actual value. Solvent accessibility was predicted using the program ASAView61 inputting the crystal structure of the unbound I domain. ΔrCαopen/closed form was calculated by measuring the Cα distance between the closed X-ray structure (PDB # 1pt6) and the averaged coordinates of each conformer of the open-bound NMR structure (PDB # 2m32).
Isothermal titration calorimetry (ITC)
Thermodynamic binding parameters for the association of Ac-(GPO)4GLOGEN(GPO)4GY-NH2 collagen peptide (synthesized by Dominique Bihan at Cambridge University and LifeTein) with wild type α1 I and E317A/α1 I were determined via isothermal titration calorimetry employing a VP-ITC (MicroCal, Northampton, MA). Protein stock solutions were dialyzed exhaustively against a buffer comprised of 5 mM PIPES, 140 mM NaCl, and 100 mM MgCl2 (pH = 7.3). The standard solutions were filtered using a 0.22 µm pore size membrane, and thoroughly degassed for 10 min. The calorimetric sample cell (1.4 mL) was filled with a 50 µM GLOGEN standard prepared in the final protein dialysate and the titration syringe (300 µL) contained a 500 µM solution of wild type α1 I or E317A/α1 I. Each titration experiment consisted of a 2.0 µL pre-injection followed by 30 consecutive 10.0 µL injections during which the reaction heats are monitored and integrated for 5.0 min. Binding isotherms were generated by recording the integrated heats normalized for α1 I concentration versus the protein:peptide ratio. A nonlinear least squares fit of the resultant profile to a single site binding model facilitates characterization of thermodynamic parameters for the protein:peptide. complex including the affinity (Ka), Gibbs free energy (ΔG), enthalpy (ΔH), entropy (ΔS), and stoichiometric ratio (n). Experimental uncertainties are expressed as fitting errors determined from least squares minimization using the Origin software program.

