Gaining weight in structural biology: Applications of mass spectrometry in protein science
After more than a century of use, the analytical tool of mass spectrometry is blooming more than ever and frequently applied in nearly all areas of the natural and life-sciences. In the last two decades mass spectrometry has become rather routine for the high-throughput analysis of peptides, forming the basis of studying proteomes in a systems-biology directed approach. The analytical power of mass spectrometry is however not limited to the analysis of peptides, and today, also intact proteins and even complete protein complexes can be analyzed, enabling MS to contribute additionally to the field of structural biology.
As illustrated nicely in this issue many complementary mass-spectrometry based approaches can be used to investigate the structure-function relationships of proteins and protein assemblies, either alone or in-concert with each other. For instance, hydrogen-deuterium exchange in combination with mass spectrometry can be used to assess which backbone hydrogen atoms are accessible to exchange, providing information on conformational changes induced in protein structures by changes in the environment and/or binding to other biomolecular entities such as drug molecules, other proteins or nucleotide sequences.
Although hydrogen/deuterium exchange has been used for decades, the rapid advances in mass spectrometric instrumentation, peptide separation and software algorithms needed to analyze the data, have made this method far more efficient in analyzing more complex systems, such as whole protein assemblies and membrane embedded proteins. Schriemer et al. describe in this issue a new “proteomics-style” workflow for analyzing HD exchange data. The usefulness of the workflow is illustrated in a conformational analysis of microtubules interacting with the dimeric kinesin MCAK that plays a role in tubular depolymerization.
Konermann et al. use MS-based HD exchange to study how proteins behave at solvent/gas interfaces testing the conformational changes occurring in myoglobin when it comes in contact with air bubbles. They argue that these surface denaturation phenomena may play an important role in therapeutic protein aggregation.
Next to HD exchange mass spectrometry, chemical cross-linking in combination with mass spectrometry has really seen a strong resurgence in the last decade, benefitting from similar advances in mass spectrometric instrumentation, novel cross-linking chemistries, peptide separation and software algorithms needed to analyze the data. Sinz and Sharon review these advances in this issue, emphasizing the potential of cross-linking (and native MS) in studying the p53 tetramer and the vinculin Arp2/3 complex.
Potier et al. combine chemical cross-linking with mass spectrometry to define a structural model and map the protein-protein interaction network of the human SAGA HAT sub-complex involved in transcription.
Another ancient approach, benefiting from the recent advances in instrumentation, sample preparation and search algorithms, is presented by Ashcroft et al., who investigate how polypeptide chains change their conformation while they are tethered to the ribosome, by using limited proteolysis in combination with mass spectrometry. Subtle differences are observed between the free and tethered forms of the studied SH3 domain containing peptide chains.
Altogether, these three approaches: HD exchange MS, cross-linking MS, and limited proteolysis combined with MS, have benefited substantially from the proteomics driven revolution in analytical technologies for peptide separation and mass analysis.
Next to these peptide-centric approaches, mass spectrometry has also made great progress over the last decade in analyzing intact proteins and protein assemblies. Analyzing proteins and protein assemblies under near physiological conditions, nowadays termed native mass spectrometry has become a powerful approach in analyzing not only the stoichiometry, but also topology and even structure of protein assemblies. In combination with tandem mass spectrometry, top-down fragmentation and especially ion mobility mass spectrometry, MS-based tools to infer structural information of assemblies as complex as whole ribosomes, viruses and nucleosomes has become possible. Ion mobility probes the collisional cross-section of gaseous particles inside the vacuum of the mass analyzer, thereby providing information on the overall shape/structure of the protein or protein assemblies.
Nowadays a body of evidence is available showcasing that at least part of the tertiary and quaternary structures of proteins and proteins assemblies is retained following the ionization process, making these cross-sections measurements valuable assets in probing the structure of biomolecules. In this issue a review by Boeri Erba et al. highlights how native MS can be used to elucidate the topology and macromolecular structures of large protein assemblies, putting special attention on how tandem mass spectrometry methods may be used to identify protein constituents from these assemblies, using top-down approaches.
Heck, Clemmer et al. use a combination of ion mobility mass spectrometry together with advanced bottom-up proteomics methods using collision induced (HCD) and electron transfer induced dissociation (ETD) to monitor in detail how the structure and conformation of human alpha defensin unfolds upon the sequential reduction of the three disulfide bridges present in this antimicrobial peptide.
Satoko et al. use a combination of native (tandem) mass spectrometry with ion mobility to study the structure of the nucleosome components H2A/H2B. By introducing charge-reducing modifications on selective arginine and lysine residues, reflecting events known to happen in vivo, they monitor changes in the structure and stability of these histone dimers by mass spectrometry.
A re-occurring important question is whether protein structures as studied in the vacuum of the mass spectrometer truly reflect the structure of the protein or protein assemblies in solution or even in vivo. Evidently, the loss of its natural environment has a substantial effect on the protein structure. Still, many studies have now revealed that the protein back-bone characteristics can often be partly retained in the gas-phase, as holds true for the quaternary organization of macromolecular assemblies. Taking this assumption as a starting point, Gross et al., Ruotolo et al. and Sobott et al. present in this issue thought-provoking results whereby they on purpose activate in the gas-phase by collisional activation the proteins and protein assemblies, to study their intrinsic unfolding. Ruotolo et al. investigate the effect of cooperative carbohydrate binding on the stability and structure of the lectin Concanavalin A by combining native MS and ion mobility mass spectrometry. Gross et al. use both electron captured induced top-down analysis of hemoglobin structural variants with native MS and ion mobility, to probe the collision induced unfolding of native-like structures of hemoglobin. Ion mobility thereby clearly reveals the overall conformational landscape of the unfolding hemoglobin proteins, whereas the top-down analysis elucidates which parts of the protein sequence unfold. Sobott et al. present work by mass spectrometry directed toward the challenging task of tackling integral membrane proteins, which they are able to analyze by reconstituting them into detergent micelles. Releasing the proteins from the micelles inside the mass spectrometer the masses and thus stoichiometries of the proteins could be established with high precision. Further collisional activation caused unfolding of these membrane protein assemblies and subsequent ejection of a highly charged monomer. Sequence tags induced by further collisional activation could be retrieved for some of these ejected monomers, highlighting how in a single set of experiments, both higher-order structures as well as protein sequences can be determined, by combining native MS with top-down MS.
Using native mass spectrometry and ion mobility as main technologies Thalassinos et al. investigate the co-occurrence of multiple stoichiometries and conformations of α1-antitrypsin that undergo various states of polymerisation due to specific point mutations. IM-MS enables them to track disease-relevant conformational behavior by studying the effects of peptide binding on the conformation and dynamics of α1-antitrypsin. Their findings were confirmed by using high resolution X-ray crystallographic and NMR spectroscopic data.
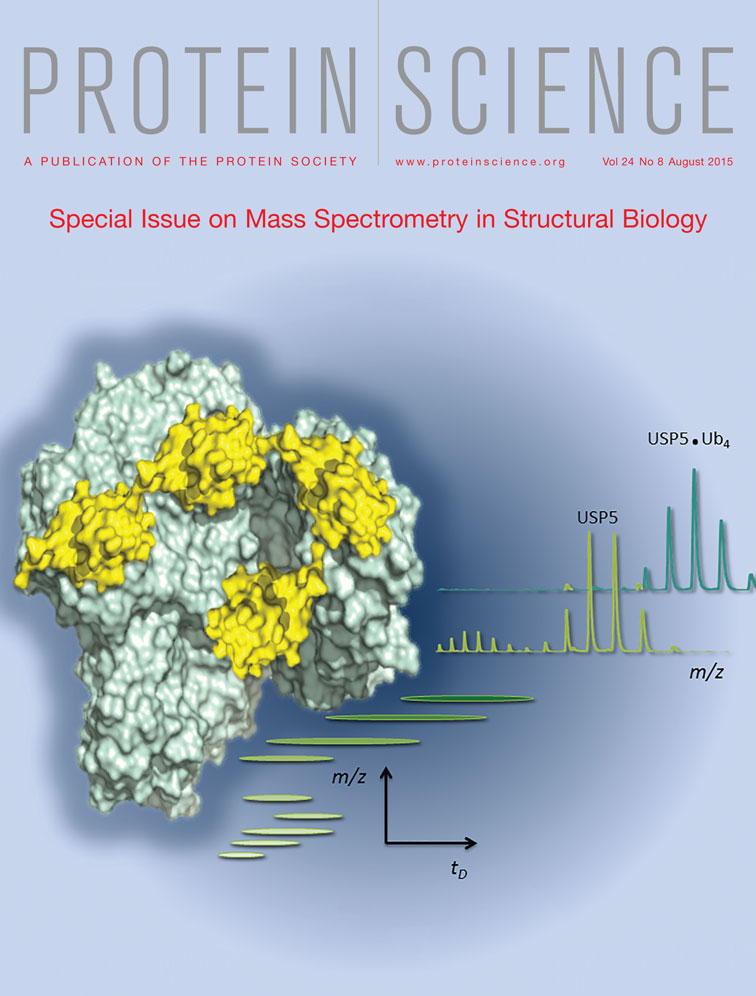
A beautiful showcase displaying how native MS and IM-MS can tackle complex biological structural questions is the work presented by Oldham et al. who investigated the interactions between the large multi-domain deubiquitinating enzyme Ubiquitin Specific Protease 5 (USP5) and its poly-ubiquitin substrates (see also cover image). Employing a C335A active site mutant of USP5 they could demonstrate by native MS that two mono-ubiquitin molecules do bind to USP5, whereas only a single tetra-ubiquitin molecule binds to the same protein. From the data on the occupied binding sites they could determine how ubiquitin binding affects the conformation of USP5 in the cases of both mono- and poly-ubiquitination.
Cianférani et al. present a study on therapeutically important antibody-drug conjugates (ADC) by combining native MS and IM-MS. They focus on the lysine-conjugated ADC trastuzumab emtansine, considered for treatment of breast cancer, that combines the anti-HER2 antibody with a cytotoxic microtubule-inhibiting maytansine derivative. Using a combination of high-resolution native MS and IM-MS they are able to monitor the drug-load on the antibody in exquisite detail, and probe the co-occurrence of positional isomers in this ADC, representing molecules with the identical drug-load, albeit conjugated at different lysine residues in the antibody. It is unlikely that any other analytical or biophysical technology can monitor the molecular complexity of ADCs in the detail described.
Overall, I am delighted to see how the work presented in this special issue of Protein Science nicely illustrates the emerging and wide-spread role of mass spectrometry in structural biology, focusing especially on the structural and functional analysis of very large dynamic proteins and protein assemblies. I hope this delight is shared by the readers, and wish you happy reading.