

Cross-nucleation of polybutene-1 Form II on Form I seeds with different morphology
Funding information: China Scholarship Council (CSC)
Abstract
Polybutene-1 (PB-1) trigonal Form I crystals with different morphologies, namely spherulitic, hedritic, and fibrous, were used as seeds to quantitatively study the cross-nucleation kinetics of the tetragonal Form II. Interestingly, a large difference in the cross-nucleation ability of the various Form I seeds was observed. The Form I fiber exhibits the fastest cross-nucleation, followed by the hedrite and eventually by the spherulite. The nucleation induction time was quantified from the evolution of the transmitted light intensity via polarized optical microscopy. The data were tested against different nucleation models, revealing that the rate-determining step for nucleation is the growth of the nucleus to attain the critical dimension, rather than the formation of the first crystalline layer in contact with the substrate. In addition, for spherulitic seeds, a meaningful dependence of cross-nucleation induction time on Form I lamellar thickness was found, with the kinetics being faster for higher original seed's crystallization temperatures. The results were interpreted assuming a free energy barrier for cross-nucleation which is a function of the mismatch between the daughter and the parent polymorphs' lamellar dimensions. This aspect, together with the calculated low values of interfacial free energy difference parameter for nucleation, suggests the possible existence of an epitaxial relationships between cross-nucleating PB-1 Form II and Form I.
1 INTRODUCTION
It is well known that many semicrystalline polymers exhibit pronounced polymorphisms,1-3 which show a strong influence on their properties. For example, isotactic polypropylene (iPP) exhibits at least three different crystal modifications, designated as α, β, and γ forms depending on the processing conditions, the chemical structure of the macromolecule, and the presence of specific additives.4-6 An earlier-nucleating crystalline polymorph can nucleate another crystalline polymorph of higher or lower thermodynamic stability, without undergoing any polymorphic transformation. This phenomenon has been named cross-nucleation and occurs on crystals sharing the same composition (although with different structure) rather than on foreign particles.7-12 Cross-nucleation between polymorphs is important for the industrial control of polymorphism and the fundamental understanding of nucleation, and it has been mainly described as a particular case of heterogeneous nucleation.
Particular attention has been paid in the past decades to heterogeneous nucleation of polymer crystals on foreign surfaces of nucleating agents (NAs) or fiber composites. An interfacial free energy difference parameter, Δσ, resulting from the formation of the crystalline layer onto the heterogeneous substrate, is often used for comparing the nucleating ability of different substrates and understanding the nucleation process on the surfaces of the heterogeneities. For example, Wang et al. studied the nucleation kinetic of iPP on the surface of different kinds of fiber and determined different values of Δσ for each system.13-15 Kawamoto et al. investigated the effect of the epitaxy of a NA on nucleation of polypropylene based on kinetic study. It was found that nucleation rate at constant undercooling increased by 60 times with decreasing Δσ/σ from 0.23 to 0.13, which confirms that Δσ is extremely sensitive for detecting the nucleating ability of a substrate.16 For what concerns cross-nucleation, although the kinetics of the process has already been studied for several systems,17-20 a detailed description based on Δσ calculation was not extensively applied.
Moreover, the effect of the substrate morphology on cross-nucleation is still poorly understood. Chen et al. studied cross-nucleation between the polymorph of “ROY” molecule, finding a meaningful effect of the nature of the seed surface on cross-nucleation rate, by comparing baked and unbaked samples.8 Tao et al. revealed that different faces of a single-crystalline seed differed in their ability to cross-nucleate. In particular, using an elongated β-phase crystal of D-Mannitol in contact with the undercooled melt, cross-nucleation of the α-phase occurred immediately if the seeds was placed side-on, while the parent β-polymorph could grow to some extent before cross-nucleation happened, in case of end-on seed orientation.11
For such a study in polymorphic polymers, polybutene-1 (PB-1) is an ideal system because it exhibits several polymorphs of known structures and known thermodynamic relations. In fact, this polymer can crystallize in Forms I/I′, II, and III.21, 22 It is well established that the Form II, commonly produced by melt crystallization, is the kinetically favored modification, but it is metastable from the thermodynamic point of view. Form II has a tetragonal unit cell with 11/3 helical conformations.23-26 Since the nucleation of the stable Form I occurs much more rarely in bulk crystallization than that of the predominant Form II, it can only be obtained through solid-solid transition from Form II. However, Form I′, described as a defective Form I with lower melting temperature, can be formed through special crystallization procedures, such as solution crystallization,27 blending with iPP,28 or copolymerization with other monomers.29-31 Actually, quiescent aging at room temperature of Form II samples is generally used for generating trigonal Form I, where the molecular conformation of the chains is that of a 3/1 helix.32-37
Given that the spherulitic seed of Form I in contact with PB-1 melt has proven to efficiently cross-nucleate the metastable polymorph,17, 38 in this work we aim at gaining some insights on the role of the substrate type on cross-nucleation. Therefore, substrates of Form I with different morphologies, namely spherulites, hedrites, or fibers, were employed to study cross-nucleation kinetics by means of polarized optical microscopy. The cross-nucleating efficiency of the various heterogeneous substrates was quantitatively compared by analyzing the “induction time” for the nucleation of Form II on the seed's surfaces, that is, the average time needed to reach a cluster of critical size. The use of appropriate heterogeneous nucleation models enabled to estimate the value of interfacial free energy difference, Δσ, for the various substrates. The observed differences were tentatively discussed on the basis of a scheme of cross-nucleation which takes into account the possible existence of a yet undisclosed epitaxial relationship between Form I and Form II, and the commensurability of the parent and daughter polymorphs' lamellar thicknesses.
2 EXPERIMENTAL SECTION
2.1 Material
The polymer studied in the present work is a butene-1 homopolymer (PB-1) with commercial-grade name of PB0110M, kindly provided by LyondellBasell. It has a weight-average molecular weight of 850 kg/mol and a polydispersity index of 6.8.
2.2 Sample preparation
Granules of PB-1 were used without any further treatment to make thin films, 30 to 50 μm thick, by compression-molding at a temperature of 180°C. PB-1 fibers used in this work were lab-spun using a capillary rheometer (RH7, Bohlin) at the Institute of Chemistry of the Chinese Academy of Science in Beijing, by the group of Prof Dujin Wang. The temperature of the barrel was 180°C and the rotation speed of the collector was set to 200 rpm.
To prepare the substrates of interest characterized by different morphologies, the thin film samples were heated up to 180°C in a Mettler FP90 hot-stage, kept at this temperature for 5 minutes to erase the previous processing history, and further cooled to the crystallization temperature. Temperatures of 90°C and 108°C were selected, in order to obtain a spherulitic and a hedritic morphology, respectively.39 After an appropriate crystallization time of approximately 4 and 270 minutes at low and high temperature, respectively, morphologies of similar size were reached in the two cases. Then the samples were quickly cooled down to room temperature and aged for more than 1 month in order to completely transform the original Form II crystals into Form I. It should be noted that, according to previously adopted procedures for cross-nucleation studies in PB-1, a dual morphology is produced.17, 38, 40 Indeed, low melting temperature crystals of Form I, formed during quenching to room temperature, are essential for having molten PB-1 in contact with crystalline Form I seeds (with higher melting temperature).17, 19
2.3 Polarized light optical microscopy
The morphology and the nucleation behavior were observed in situ by a Leica DMLP optical microscope under crossed polarizers, equipped with a computer-controlled digital camera (Optika B5). The thermal treatments were applied by a calibrated Mettler Toledo FP82HT hot-stage. To investigate the nucleation on the substrates of spherulite and hedrite, the Form I films were heated to different melting temperatures (Th) at 20°C/min, and kept for 5 minutes to melt completely Form II crystals or Form I crystals with lower thermal stability, and subsequently quenched to Tc for performing the isothermal crystallization (Figure 1A).

For the experiments with the fiber substrate, these thin PB-1 films were first heated to 180°C and kept at this temperature for 5 minutes to erase the previous processing history, then they were cooled down to different melting temperatures (Th). The PB-1 fibers, containing Form I crystals, were then manually introduced in the film at this temperature. After a second annealing step at Th, to relax the induced stresses, the samples were cooled down to the selected crystallization temperature (Tc) (Figure 1B). The described thermal protocols are presented in Figure 1.
3 RESULTS AND DISCUSSION
3.1 Determination of nucleation induction time via light intensity measurements during crystallization
Figure 2 shows an example of the time-resolved images obtained during isothermal crystallization, and explains the method used for induction time determination. Nucleation of Form II crystals occurs at every place on the side of the original Form I spherulite, forming a sort of crystalline “corona” around it (Figure 2A). In order to accurately detect the induction time, that is, the time at which the Form II cross-nuclei have reached a critical size and the transcrystalline growth proceeds, the increase of the light intensity in the sample due to the formation of birefringent Form II crystals is exploited. A circular “Region of Interest,” centered in the middle of the nucleating spherulite, is selected on the image analysis software, and the mean light intensity in the selected area is then calculated for each acquired frame. By plotting this intensity as a function of time (Figure 2B), it is possible to unambiguously determine the time at which it starts to increase from its initial value, that is, the induction time. Further details on the analysis method can be found in our recent work.41 The exact value of the induction time is determined by the intersection point between two fitting lines: one in the time region of the initial plateau value and the second in the first growth step part. An example of the analyzed data is reported in Figure 2B.

3.2 Cross-nucleation of PB-1 Form II on Form I seeds with different morphology
Figure 3 shows some representative optical micrographs of PB-1 crystallized on the surface of Form I substrates with different morphologies: spherulite, hedrite, and fiber. All the samples were crystallized isothermally at 105°C, after melting the original Form II/Form I crystals at 124°C. The thermal treatment at relatively high temperature ensures that the remaining seeds are composed of Form I only, while any crystalline memory of possible residual Form II crystals (ie, self-nucleation) is erased.40 In all the three morphologies, a clear transcrystalline layer (TCL), indicative of a very high “linear density” of nucleation sites, develops with time. The TCL13, 14, 42, 43 is a consequence of lack of lateral separation between the individual growing spherulites on the nucleating surface, causing the crystals to grow exclusively perpendicularly to it. It is apparent that the nucleating efficiency of the three substrates is particularly high; however, some subtle difference among the three can be captured. In fact, while individual splaying spherulites can be somehow recognized in Figure 3A,B (with spherulitic or hedritic Form I seeds), this is not the case for the crystals nucleating on the fiber surface, due to the extremely high nucleation density.

However, the different cross-nucleating ability of the various substrates can be more clearly deduced by comparing the time evolution of the morphology, at the same crystallization temperature. The second column of Figure 3 reports for each case the time at which the first cross-nucleation event could be observed (highlighted by the red circles). It can be seen that a substantially shorter time is required for cross-nucleation to occur on the fiber substrate, with respect to the spherulitic or hedritic ones. In the last images of Figure 3A-C, the TCL has grown to a similar thickness in all the three morphologies, being the growth rate the same in the three experiments, the earlier onset of growth, that is, shorter induction time, for the fiber sample is confirmed.
During primary nucleation, a particular molecular arrangement must be fulfilled to allow the formation of the new phase. In the case of polymer crystallization from the melt, the chain segments must approach themselves to distances commensurate to those characteristics of the crystalline unit cell and must possess the right conformation, as the crystal symmetry imposes. The chain segments with those attributes form a crystalline aggregate which varies the system free energy ΔG. Considering for the sake of simplicity a spherical nucleus of radius r, a maximum in ΔG(r) exists which corresponds to the critical size (r*) that the new crystal must attain in order to be able to spontaneously grow into a crystal. Clusters smaller than r* are called subcritical nuclei or embryos, they are unstable and tend to disappear from the system. Aggregates that exceed r* are called supercritical nuclei and can continue to grow because their growth is thermodynamically favored.44
As the magnitude of the energy barrier for nucleation depends inversely on the undercooling, that is, on the distance between the crystallization and melting temperature, experiments were performed at different crystallization temperatures. Figure 4A reports the example of evolution of light intensity, extracted from the optical micrographs, as a function of time during crystallization at different temperatures for the fibrous Form I substrate. The time of formation of the critical nuclei, represented by the light intensity induction time, gets shorter and shorter with decreasing Tc for any of the considered substrates. In particular, while the differences between spherulitic and hedritic Form I seeds are relatively small (data are reported in Figure S1), the high cross-nucleation efficiency of the Form I fiber is apparent by comparing the time scale of the x-axis (Figure 4A). A clearer comparison between the three seeds is offered in Figure 4B, which considers the increase in transmitted light at the same crystallization temperature for the three different systems. The chosen condition is the same reported in the micrographs of Figure 3. A difference of about a factor 0.8 in the induction time can be already appreciated between the spherulitic or hedritic seeds, while Form I fibers substrate can nucleate Form II about two to three times faster than Form I spherulite.

In order to apply a model-based comparison of the cross-nucleation ability associated to different Form I morphologies, the values of the determined induction times as a function of temperature are reported in Figure 5. The order of efficiency deduced from Figures 3, 4, and S1, that is, fiber > hedrite > spherulite, is confirmed in the whole range of explored undercooling. As expected, a strong dependence of nucleation kinetics on the crystallization temperature is observed, with induction times increasing exponentially approaching the melting point of Form II.

It should be pointed out that both spherulitic and hedritic Form I substrates were prepared in situ in the sample, by melt crystallization, while fibrous Form I seed was introduced into the matrix melt manually. The insertion process necessarily involved shear stresses applied to the PB-1 melt. Given the fact that flow-induced nucleation in fiber-pulling experiments is well documented, and is known to have long-lasting effects,45 the evaluation of its importance in the present case is mandatory, in order to understand whether a fiber-like Form I really possesses the highest cross-nucleation efficiency.
The annealing times were varied in a wide range, according to the known long lifetime of PB-1 flow-induced precursors.45 Figure S2 reveals that the change of the induction time, with melt annealing time, if any, is safely negligible and cannot account for the large difference observed for the case between the nucleation time of Form II at the interface with the fiber and with the hedrite of Form I (around 140 seconds).
To further confirm that nucleation of Form II on top of Form I fibers is not dominated by shear-induced effects, the same experiment, including the fiber introduction step, using a completely different fiber, made of iPP, was performed. Since the elastic moduli and size of the two fibers are not largely different, similar shear stresses are expected to be generated by the fiber introduction procedure in the two cases. The induction times observed for nucleation on iPP and Form I PB-1 fibers are compared in Figure S3. Very different nucleation kinetics of Form II are observed on the two fibers. In particular, nucleation on iPP fiber occurs more slowly than cross-nucleation on Form I PB-1 fiber. This observation further supports our previous conclusion: the effect of shear-induced nucleation due to the sample preparation procedure is not meaningful. In the opposite case, a similar kinetic of nucleation would be observed for the two fibers. Thus, the observed faster nucleation promoted by the Form I PB-1 fiber, with respect to the other PB-1 substrates made of the same crystalline polymorph but possessing different morphologies, is confirmed.
3.3 Crystal growth of PB-1 Form II on Form I seeds with different morphology
In order to exclude any possible difference between the growth rates of Form II crystals in the different samples, and in view of obtaining surface energy data for the subsequent estimation of the nucleation barrier, the growth kinetics in the different samples was determined by following the time dependence of the TCL thickness.
 (1)
(1)The constants that appear in Equation (1) are: G0 is a temperature-independent parameter, U* is the activation energy related to the transport of chain segments across the phase boundary, R is the gas constant, T ∞ is the temperature below which all motions associated with viscous flow cease, β is a constant characterizing the regime of growth, b0 is the thickness of each newly formed layer, σ and σe are the lateral and fold surface energies, respectively; Tm is the equilibrium melting temperature, k is the Boltzmann constant, Δh0 is the melting enthalpy, and f is a correction factor which takes into account the change of melting enthalpy with crystallization temperature.
Figure S5 shows the trend of log G + U*/R(T−T ∞) as a function of 1/(TΔTf). The linear fit of the growth rate data with the L-H model with a single slope in the whole undercooling range implies that only one regime is active between 100°C and 110°C. This corresponds to regime I, in agreement with the results of Monasse and Haudin.46 On the other hand, it is apparent that the TCL of PB-1 Form II grows in the melt with the same rate, independently from the exact substrate on which it nucleates. Therefore, it can be concluded that the observed differences in the kinetics of light intensity evolution among the different Form I seeds are not related to the growth stage, but can correctly be attributed to the cross-nucleation kinetics only.
 (2)
(2) (3)
(3)The relationship between the growth rate values of the TCL, calculated through the thickness vs time measurements and the light intensity method, has been further explored. Figure 6 shows the growth rate values calculated at different Tc from TCL thickness analysis (Figure S4), as a function of the parameter Aν, defined as the square root of the fitting slope of the linear part in the plot of the light intensity method (Figure S6). From Equation (3), we can derive that A is a constant equal to (KZ)1/2. If the applied model is correct, a linear relationship between these two independently derived quantities should be obtained. As can be seen in Figure 6, the growth rate data for all the different substrates fall on the same line. Therefore, we can conclude that this model for the data analysis is validated, analogously to our previous work on heterogeneous nucleation of polypropylene,41 and that the parameters K and Z are independent from the morphology of the Form I seeds.

3.4 Model analysis for cross-nucleation of PB-1 Form II on Form I seeds
 (4)
(4) (5)
(5) (6)
(6)By plotting ln I + U*/R(T−T ∞) vs 1/T(ΔTf)2, a linear fitting can be obtained and the slope (K i) of the line is proportional to σσeΔσ. In order to determine Δσ, one also needs to measure the growth rate at different crystallization temperatures and determine the value of the σσe product from its temperature variation. In fact, according to L-H growth model (Equation 1), a plot of ln G + U*/R(T−T ∞) vs 1/TΔTf yields a straight line with a slope K g, proportional to σσe. From these two separate sets of experiments, Δσ is obtained. Assuming from the literature an activation energy U* of 6.28 kJ/mol, a bulk enthalpy of fusion Δh0 of the Form II of 56 J/cm3 and a molecular stem width b0 of 7.45 × 10−8 cm,49 three very close values of the term σσe has been calculated for cross-nucleation on spherulitic, hedritic, and fibrous Form I seeds: 56.07 erg2 cm−4, 56.63 erg2 cm−4, and 53.41 erg2 cm−4, respectively.
 (7)
(7)Therefore, by relating the nucleation rate and the induction time an efficient way to obtain Δσ is provided.
In Figure 7 Ishida's model is applied to the measured cross-nucleation induction times for PB-1 nucleated onto different Form I seeds. A good linearity of the data is observed, allowing us to perform a linear fitting and derive the slope K i, that is, calculate the quantity σσeΔσ. Combining it with the σσe calculated from K g obtained from the Lauritzen-Hoffman plot (Figure S5), an estimate of Δσ for the spherulite, the hedrite, and the fiber substrate can be derived: 0.294 erg cm−2, 0.301 erg cm−2, and 0.291 erg cm−2, respectively.

As judging from the obtained values, the model of Ishida is not capable of capturing significant differences between the nucleating efficiency of the different substrates. In fact, the lines of Figure 7 are practically parallel to each other, indicating that the same cross-nucleation barrier would be obtained for the different Form I seeds. Clearly this conclusion cannot account for the differences experimentally observed in the induction time values of each system, given the negligible difference in the growth rate.
 (8)
(8) (9)
(9) (10)
(10)
As can be noticed, Q includes the interfacial free energy difference (Δσ).
Figure 8 shows the evolution of ln(t iΔT) − U*/R(T−T ∞) as a function of 1/TfΔT for PB-1 matrix crystallized in contact with Form I seeds with different morphologies. The analysis of the cross-nucleation induction time data with a model which expresses the nucleation barrier taking into account the formation of several crystalline layers, up to the attainment of critical dimensions, can correctly capture the different nucleating ability of the various substrates. In fact, the lines display a different intercept with the y-axis, given by different Δσ. However, its absolute value cannot be obtained due to the unknown constant C.

Therefore, in order to correctly quantify the nucleating activity of each substrate, a new approach, consisting in the use of the sum of Equations (8) and (9) to fit the experimental induction time data, is required. In this way, a better identification of the crystallization temperature window in which each one of Muchova's models (th and ts) is valid, can be obtained. The model describing the total induction time as the sum of the individual steps is referred as “detailed model” in the following.
Indeed, the description of the total induction time by the detailed model allows us to estimate in which range of temperature each step is dominant and what is its exact contribution to the overall induction time at a given crystallization temperature. In Figure 9A and Figure S7, the contributions of first and further layers models to the detailed model are shown for each system with different PB-1 Form I seeds. It is clear that the contribution of the further layer model (ts) is much larger than that of the first layer model (th) at higher temperatures (>100°C). This means that the data in the test temperature range follow very closely the trend described by the ts model alone, that is, the rate determining step in the attainment of the nucleus critical size is the addition of several crystalline layers. On the contrary, for lower crystallization temperatures (<87°C), the formation of the first crystalline layer is enough for the nucleus to attain the critical size.

Figure 9B shows the curves obtained by fitting the experimental induction time data with the detailed model equation for the three PB-1 Form I substrates. The parameter A1 from Equation (8) was obtained using crystallization induction time data of bulk PB-1, given that nucleation is likely controlled by the formation of the first layer onto substrates of low nucleation efficiency and at high undercooling.41 A value of 0.005 second could be determined and used in the subsequent fit of the data of interest for all the substrates. The resulting values of the unknown parameters from the sum of Equations (8) and (9) (A2, Δσspherulite, Δσhedrite, and Δσfiber) are 0.002 second, 0.295 erg cm−2, 0.225 erg cm−2, and 0.105 erg cm−2, respectively. Therefore, the Δσ values obtained for the three substrates agree with the experimental trend of nucleation kinetics.
Given the results discussed above, it can be concluded that the kinetic of nucleation cannot be estimated by modeling a single step only, but rather by considering the two steps simultaneously. Figure S8 shows the plot of the curves relative to the first layer model (th) and the further layer model (ts) obtained during the fitting of the detailed model for the different Form I seeds with varying morphologies. Within the range of experimental temperature, the differences between th and ts curves at the same crystallization temperature increases in the order: fiber > hedrite > spherulite. This order mirrors the influence of the formation of the first crystalline layer on the nucleation barrier, that is, the lower the Δσ, the less important becomes the first step in determining the overall induction time. In view of interpreting the observed effect of seed's morphology on cross-nucleation, it is worth to briefly consider the absolute values of the obtained free energy difference parameters. For all the considered substrates, extremely low values of Δσ were found, in comparison to the systems for which most of the literature data are available, that is, polymer/fiber composites, with values ranging from 4 to 20 erg cm−2.42, 51 On the other hand, such low values are comparable to those obtained in particular cases, such as those of ultrahigh molecular weight polyethylene fibers in polyethylene or polycaprolactone matrices,48, 52 for which epitaxial nucleation is obvious or well known.53 Thus, particularly favorable interactions between crystallizing Form II and Form I seeds must be in place. It is conceivable that cross-nucleation in PB-1 is governed by either “true” crystallographic epitaxy between the two structures, or at least by a “soft epitaxy” mechanism related to a matching between the topographical details of parent and daughter crystals at the nanoscale.
4 DISCUSSION
Through the above study of the cross-nucleation of PB-1 Form II on Form I substrates, it has been shown that the nucleation activities are significantly affected by the substrate type of seed. Given that all the substrates and the crystallizing polymer have exactly the same chemistry, the different nucleating efficiencies should be attributed to peculiar physical interactions rather than chemical ones. The crystalline substrate possesses a multi length-scale structure ranging from molecular to mesoscopic scale. It is not yet clear which one of these length scales has the largest impact on cross-nucleation.
In most cases of nucleation induced by the presence of solid fibers in composites, the fiber surface topography was proposed to have a determining role in nucleation kinetics. This was mainly attributed to two reasons. On one hand, thermal stresses from temperature change at the coarser fiber interface might induce local orientation of polymer chain segments, providing efficient seeds for nucleation. With respect to the smooth surface, such thermal stresses are expected to be larger at deep “valleys.”54 On the other hand, for forming a viable nucleus, the free-energy barrier is always larger on a flat surface than that in a groove, due to the possibility of secondary or tertiary nucleation.42 In the present case, the surface topography of the used seeds with different morphologies is not easily accessible, especially for spherulitic and hedritic seeds, since they are embedded in the crystallizing PB-1 itself. Furthermore, it is difficult to explain the effect of surface topography on the nucleation by considering the degree of roughness alone.51 Dalnoki-Veress and colleagues55 studied the role of the interface in inducing crystal nucleation by using isotactic polystyrene (i-PS) as a substrate for crystallizing poly(ethylene oxide) (PEO) droplets. They found a larger surface roughness and a higher nucleation temperature for i-PS film isothermally crystallized at 175°C with respect to the same film crystallized at 185°C. However, these results are not in agreement with the ones presented hereby, since cross-nucleation kinetic onto Form I spherulite, formed during isothermal crystallization at 90°C, is slower than that on Form I hedrite (isothermally crystallized at 108°C). Therefore, assuming a similar dependence of the crystalline seed roughness in the two cases, other different factors should be considered in order to explain the cross-nucleation efficiency of the different PB-1 Form I substrates.
On the other hand, the role of molecular epitaxy should not be overlooked. For instance, Damman et al.56 showed that molecular epitaxy played a greater role in the enhancement of nucleation of a small molecule, in comparison to surface topography. In the case of cross-nucleation between polymorphs, the role of epitaxy is controversial. For example, in the solution crystallization of a steroid, cross-nucleation of the metastable polymorph on the stable one can occur both with and without preferred mutual orientation between the two structures, depending on the solvent mixture.57, 58 In polymers, cross-nucleation of the monoclinic phase on trigonal crystals via epitaxial mechanism has been highlighted in single crystals,59 but not in melt-crystallization.60, 61 Focusing on the cross-nucleation of PB-1 Form II on Form I, the existence of a specific epitaxial relationship between the two polymorphs has not been disclosed yet.
For the sake of the following discussion, we assume that a preferred mutual orientation of the chains in the two structures (PB-1 Form I and Form II) must exist at the cross-nucleation point. This can result from a strict crystallographic epitaxy or from a more general “soft epitaxy” mechanism. Several cases of epitaxy between semicrystalline polymers have been studied in the literature.62, 63 It has been recognized that, since the crystalline substrate has itself a lamellar morphology, the requirement of lattice match between the two structures is not enough to allow the occurrence of epitaxy. In fact, because of the limited dimensions of the substrate crystals, a matching between the lamellar sizes of the two polymers must also exist.64 A paradigmatic example of this constraint is found in the system isotactic polypropylene/polyethylene. Epitaxy of the crystallizing polyethylene onto the semicrystalline polypropylene substrate can only occur for crystallization temperatures below 123°C.65, 66 Therefore, it is of interest to test the possible dependence of PB-1 Form II cross-nucleation kinetics on the lamellar thickness of Form I seeds. Figure 10 shows the induction time of cross-nucleation for Form II on spherulitic Form I seeds originally crystallized at different temperatures, as a function of the crystallization cross-nucleation temperature. Data for the hedritic morphology (sample crystallized at 108°C) are included as well. As can be seen, the induction time clearly decreases, and thus the efficiency of cross-nucleation increases, with increasing the spherulite substrate preparation temperature, in the whole range of explored supercooling. Since it is generally known that lamellar thickness is proportional to the crystallization temperature, it can be deduced that the nucleating ability of the substrates depends on the Form I seed's lamellar thickness, rather than on the specific orientation of the Form I lamellae (edge on vs flat on in spherulitic or hedritic seeds, respectively).

Given the above findings and the apparent analogy between polymer-polymer epitaxy and cross-nucleation, we can tentatively interpret the effect of seed morphology on nucleation on the basis of the “template model” proposed by Phillips and colleagues to link epitaxy and secondary nucleation.64 In this model, the author recognized that for the epitaxial deposition of a polymer onto another one, the crystal dimension of the substrate in the direction characteristic of the lattice match must be larger than the critical stem length of the crystallizing polymer's secondary nucleus. The proposed scheme is adapted to the present case of PB-1, as sketched in Figure 11.

Considering a given crystallization temperature, the critical stem length of the Form II secondary nucleus is indicated as
l, while the generic Form I seed is characterized by a crystalline lamellar thickness and amorphous layer thickness 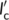 and
and 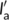 , respectively. The deposition of Form II stems occurs at an angle θ with respect to the lamellar normal of the Form I substrate crystal, as, dictated by the epitaxial lattice matching (unknown at present). Therefore, it is possible to infer variations in the free energy barrier for cross-nucleation on different substrates, by comparing the magnitude of
, respectively. The deposition of Form II stems occurs at an angle θ with respect to the lamellar normal of the Form I substrate crystal, as, dictated by the epitaxial lattice matching (unknown at present). Therefore, it is possible to infer variations in the free energy barrier for cross-nucleation on different substrates, by comparing the magnitude of 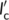 and
l in the lattice matching direction, depending on seed's formation temperature and actual crystallization temperature.
and
l in the lattice matching direction, depending on seed's formation temperature and actual crystallization temperature.
Given the defined angle between the polymorphs' chain directions, the maximum parent crystal dimension available is 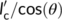 . If the critical stem length of Form II at the crystallization temperature is shorter than
. If the critical stem length of Form II at the crystallization temperature is shorter than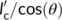 , a nucleus is easily generated onto the surfaces of Form I crystalline substrate. This is certainly valid for the hedritic morphology (Figure 11A), since the seed is originally crystallized at 108°C, at the high end of the investigated cross-nucleation temperature range (100°C-110°C). Thus, the lamellar thickness of the Form I seed is expected to be typically larger than the critical stem length of the cross-nucleating Form II.
, a nucleus is easily generated onto the surfaces of Form I crystalline substrate. This is certainly valid for the hedritic morphology (Figure 11A), since the seed is originally crystallized at 108°C, at the high end of the investigated cross-nucleation temperature range (100°C-110°C). Thus, the lamellar thickness of the Form I seed is expected to be typically larger than the critical stem length of the cross-nucleating Form II.
On the contrary, when
l is larger than , a portion of the deposited macromolecular chain segment will lay on the amorphous phase. For that part of the nucleus, no decrease in free energy associated with the formation of the related crystal volume is expected, while a lateral surface free energy penalty is still generated. This implies that the nucleation energy barrier will be increased by an amount proportional to
, a portion of the deposited macromolecular chain segment will lay on the amorphous phase. For that part of the nucleus, no decrease in free energy associated with the formation of the related crystal volume is expected, while a lateral surface free energy penalty is still generated. This implies that the nucleation energy barrier will be increased by an amount proportional to 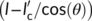 , and up to a maximum value proportional to
, and up to a maximum value proportional to 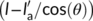 when the secondary nucleus is formed on two adjacent lamellae of the seed.64 This condition is likely to be encountered in the case of the spherulitic Form I seed, since the parent polymorph lamellar thickness is smaller than that of cross-nucleating Form II crystals (Figure 11B). This is because the temperature used to prepare the spherulitic substrates ranges between 75°C and 90°C, at least from 10°C to 20°C lower than the temperatures used for the cross-nucleation experiments. Therefore, the kinetics of cross-nucleation on the surface of spherulitic Form I substrate is slower with respect to the one on the hedrite, due to the higher nucleation energy barrier. Furthermore, within the spherulitic seeds crystallized at different temperatures, the increase in nucleation barrier due to mismatch between From II and Form I lamellae will be the highest for the lowest seed's lamellar thickness.
when the secondary nucleus is formed on two adjacent lamellae of the seed.64 This condition is likely to be encountered in the case of the spherulitic Form I seed, since the parent polymorph lamellar thickness is smaller than that of cross-nucleating Form II crystals (Figure 11B). This is because the temperature used to prepare the spherulitic substrates ranges between 75°C and 90°C, at least from 10°C to 20°C lower than the temperatures used for the cross-nucleation experiments. Therefore, the kinetics of cross-nucleation on the surface of spherulitic Form I substrate is slower with respect to the one on the hedrite, due to the higher nucleation energy barrier. Furthermore, within the spherulitic seeds crystallized at different temperatures, the increase in nucleation barrier due to mismatch between From II and Form I lamellae will be the highest for the lowest seed's lamellar thickness.
While this model is rather intuitively applicable when both the parent and daughter polymorphs exhibit a lamellar structure, such as in the case of PB-1 Form II spherulites nucleating on Form I spherulites or hedrites, the situation is not straightforward for the cross-nucleation on the Form I fibrous seeds. In fact, semicrystalline fibers are composed of several long fibrils constituted of smaller oriented crystallites interconnected via tie-chain in the amorphous phase (Figure 11C). The size of these crystalline blocks along the polymer chain direction might be comparable to the lamellar thickness of the hedrite sample, as judged by the similar melting temperatures recorded by differential scanning calorimetry (see Figure S9). However, under the assumption that the same cross-nucleation model would indifferently apply to both morphologies, it must be deduced that the specific arrangement of crystalline and amorphous Form I chains at the nanoscale in the fiber sample is more favorable to cross-nucleation. We can speculate that the very similar lamellar thickness between the crystallizing Form II and the hedritic Form I seed would still result in a certain energy penalty to the nucleus formation, due to the high probability of Form II stem deposition onto the chain folding region. On the other hand, the lack of perfect lateral correlation between the crystallite position in the fibers likely ensue in the availability of a larger Form I crystalline substrate along the matching direction with the cross-nucleating Form II (as schematized in Figure 11C). Moreover, the preferential extended chain polymer conformation in the fibrillar crystals, with a reduced presence of chain folds at the lamellar edges, might possibly cause a less severe energy penalty for the depositing Form II stem, with respect to the hedritic chain folded morphology.
In conclusion, the cross-nucleation of PB-1 Form II onto Form I substrates must involve the necessary requirements for the formation of a Form II secondary nucleus of critical size. The differences in cross-nucleation rate among the seeds with different morphologies can be ascribed to the different crystal dimensions of the Form I substrates in comparison to the lamellar thickness of the nucleated Form II, projected along a certain direction. This mechanism has been rigorously applied in the literature65, 66 to the epitaxy between different polymer pairs. This result suggests that it might also describe the case of PB-1 cross-nucleation, although a detailed epitaxial relationship between Form I and Form II is still not known.
5 CONCLUSION
In the present work, the cross-nucleation behavior of Form II on seeds of Form I possessing different morphologies was investigated. It was found that the cross-nucleation ability of Form I seeds toward Form II varies with substrates. In particular, the cross-nucleation efficiency decreases in the order: fiber > hedrite > spherulite. On the basis of a quantitative analysis of the induction time, the cross-nucleation energy barrier was successfully determined, revealing that the rate determining step in the cross-nucleus formation is the attainment of the critical size, rather than the formation of the first crystalline layer on the substrate. The observed different cross-nucleation efficiencies of the seeds are attributed to differences in the Form I lamellar thickness, and to its mismatch with the Form II cross-nucleus critical size. This is in agreement with a previously developed model which describes polymer-polymer epitaxy, and suggests that a defined mutual orientation between the two polymorphs' structures might be required at the cross-nucleation point.
ACKNOWLEDGMENTS
W.W. thanks the China Scholarship Council (CSC) for funding his PhD scholarship. D.C. and W.W. acknowledge Prof Shouke Yan and Prof Giovanni Carlo Alfonso for insightful discussions.

