

Desorption of myo-inositol hexakisphosphate and phosphate from goethite by different reagents
Abstract
Inositol phosphates are abundant organic phosphates found widely in the environment. The sorption and desorption of organic phosphate (Po) are important processes in controlling the mobility, bioavailability and fate of phosphorus (P) in soil and sediment. The desorption characteristics of myo-inositol hexakisphosphate (IHP) and inorganic phosphate (Pi) from goethite were studied by pre-sorption of IHP or Pi followed by desorption by KCl, H2O, and citrate. Batch experiments and in situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy were used to investigate the desorption of IHP/Pi. The desorption percentage of IHP/Pi by citrate was much higher than that by H2O/KCl. The desorption of P by citrate was mainly achieved through ligand exchange, and the desorption increased with decreasing pH. Desorption by H2O was slightly greater than that by 0.02 M KCl because the electrostatic repulsion between the P molecules is larger in H2O. Due to the higher affinity of IHP for goethite than that of Pi, the maximum desorption of IHP was lower than that of Pi. Desorption curves (desorption concentration in solution vs. sorption density) of IHP or Pi on goethite by KCl or H2O was well fitted by an exponential equation, while those by citrate were well fitted by a linear equation. The desorption amounts of P in the first cycle account for more than 58% of the total desorption followed by substantial decreases in the second and third cycles. There was a re-sorption of Pi from solution in the late stage of desorption by KCl and H2O, resulting in a sharp decrease in desorption. Re-sorption of IHP did not occur, which is probably due to its poor diffusion into goethite. The initial desorption rate of Pi with KCl and H2O decreased with increasing pre-sorption time, whereas that of IHP was opposite. This study indicates that strong sorption on and weak desorption of IHP from iron (hydr)oxides may explain the accumulation of IHP in soils.
1 Introduction
Phosphorus (P) is a plant nutrient and also a major contributing factor to eutrophication and non-point source pollution (Conley et al., 2009). Adsorption, desorption, dissolution, and precipitation reactions of P at mineral interfaces affect the mobility, bioavailability, and dynamics of P (Vance et al., 2003; Olsson et al., 2010; Ruttenberg and Sulak, 2011; Yan et al., 2014a). In addition to inorganic phosphate (Pi), organic phosphate (Po) is an important P pool commonly involved in biological processes (Turner et al., 2005). Sorption and desorption behaviors of both Pi and Po on soil mineral surfaces need to be considered (Olsson et al., 2010).
Goethite is one of the most abundant naturally occurring iron oxides in soils and has great interaction with IHP. IHP is sorbed on goethite most probably by forming inner-sphere surface complexes, in which four phosphate groups in every IHP molecule bind on goethite, while the other ones remain free (Ognalaga et al., 1994; Celi et al., 2001). However, Johnson et al. (2012) proposed that adsorption of IHP on goethite occurred by outer-sphere complexation and hydrogen bonding played an important role. The amount of desorbed IHP from goethite increased with increasing pH and the number of desorption cycles, and citrate solution desorbed a smaller amount of IHP when compared with Pi (Martin et al., 2004). These findings suggest that IHP shows a lower mobility than Pi. Other soil components, such as ferrihydrite (Celi et al., 2003), hematite (Giaveno et al., 2008; Yan et al., 2014c), gibbsite (Giaveno et al., 2008; Ruyter-Hooley et al., 2015), amorphous aluminum hydroxide (Guan et al., 2006; Yan et al., 2014a), and boehmite (Yan et al., 2014b) also had great interaction with IHP. The sorption of IHP on soil sorbents resulted in the accumulation of IHP in soils (Turner et al., 2005).
The sorption of IHP on mineral surface affects the availability of IHP (Gerke, 2015a). It was concluded that the strength and extent of IHP binding to soil components were more important than the persistence of phytase activity in controlling the release of phosphate (Giaveno et al., 2010). Martin et al. (2004) studied the desorption and plant availability of IHP sorbed on goethite and found that ryegrass that received P as IHP showed limited growth and symptom of P deficiency. It was reported that ryegrass was able to utilize IHP when it was in solution but not when it was strongly bound to soil minerals (Dalal, 1977). The co-exudation of plant organic anions and phytase was proposed as an approach to overcoming the limited ability of plants to utilize insoluble IHP in soils (Giles et al., 2012, 2014; Lung and Lim, 2006). Therefore, desorption of IHP from mineral surfaces or production of soluble IHP by organic anions was necessary for plant utilization or the activity of phytase enzymes (Gerke, 2015b).
To gain more insights into the desorption behavior of inositol phosphate, the desorption of IHP adsorbed on goethite at pH 5.0 in 0.1 M KCl by three desorbing reagents (0.02 M KCl, H2O, and 0.02 M citrate at pH 6) was investigated in this study by using a combination of quantitative batch desorption measurements and attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. KCl and H2O were used as desorbing reagents to simulate the soil solution with varying ionic strengths. To simulate the effect of low-molecular weight organic acids on the desorption of IHP/Pi, citrate was also chosen. Parallel experiments were carried out with Pi for comparative purpose. The effects of the types of desorbing reagents, desorption cycles, pre-sorption time and density, and citrate-dependent pH on desorption reaction of IHP were examined.
2 Material and methods
2.1 Reagents
Solutions and Fe oxide suspensions used in this study were prepared with analytical reagent grade chemicals and ultra-pure water (ρ > 18 MΩ cm). Dipotassium IHP (≥ 95% purity) was obtained from Sigma-Aldrich (St. Louis, Missouri, USA). Stock solutions of IHP and KH2PO4 were prepared and stored at 4°C. Hydrolysis of IHP in the stock solution was monitored by measuring the Pi level before each use.
2.2 Goethite preparation
Goethite was synthesized according to the method of Cornell and Schwertmann (2003). The obtained goethite was analyzed by powder X-ray diffraction (XRD; Bruker AXS Gmbh, Karlsruhe, Germany) and transmission electron microscopy (TEM; Phillips CM12, Philips, Netherlands) to check the crystal structure, morphology, and particle size. The specific surface area (SSA) of the synthesized goethite was measured by N2 adsorption on an Autosorb-1 standard physical adsorption analyzer (Quantachrome Autosorb-1, Florida, USA).
2.3 Zeta potential analysis
The point of zero charge (PZC) of goethite in 0.001 M KCl was determined with a zeta (ζ) potential analyzer (Malvern Zetasizer ZEN 3600, Worcestershire, U.K.). Changes in ζ potential of goethite due to P sorption over time were also examined. Goethite suspensions (0.05 g L−1) were prepared with 0.87 μM IHP or 4.35 μM Pi in 0.001 M KCl. The pH of suspensions was kept at pH 5 during the experiments with small additions of 0.1 M HCl or KOH. From the stirred suspensions, aliquots were collected at selected time intervals (1, 3, 6, 12, 24, 48, 72, 96, 168, and 336 h) for ζ potential analysis.
2.4 Sorption and desorption isotherms
Goethite suspension (5 g L−1), dispersed in 0.1 M KCl, was acidified with 0.1 M HCl to a final pH of 5.0 ± 0.05 and purged with N2 for 24 h to remove CO2. During this period, the pH was regularly checked and re-adjusted to pH 5. From this suspension stock aliquots of 10.0 mL (corresponding to 50 mg goethite) were added to a series of 10.0 mL P-containing solutions (14.5–145 and 43.5–435 μM for IHP and Pi, respectively) at pH 5.0 in 0.1 M KCl. The suspensions were equilibrated by shaking at 200 r min−1 and 25°C in the dark. During equilibration, the pH of each batch sample was measured after 6, 24, and 30 h and adjusted to pH 5.0 ± 0.05 with 0.1 M HCl (or KOH). After 30 h the suspensions were centrifuged at 9500 × g for 15 min. The supernatants were decanted and filtered through a 0.22-μm Millipore membrane and analyzed for obtaining the equilibrium P concentrations and calculating the sorption density of P based on the difference in concentration before and after P sorption.
The sediments were washed with 0.1 M KCl at pH 5.0 three times to remove the residual P and used as starting material for the 24 h desorption experiments. To achieve desorption, 10 mL of H2O, 0.02 M KCl, or 0.02 M citrate at pH 6.0 were added to the sediments and the mixtures were shaken at 200 r min−1 for 24 h. The resulting suspensions were centrifuged and the supernatants were filtered through a 0.22-μm membrane for P analyses and calculation of the desorption amounts. Three sequential desorption cycles were run for each sample, repeating the procedure described above.
2.5 Desorption kinetics
A series of goethite suspensions of 5 g L−1 was equilibrated at pH 5 in 0.1 M KCl under N2 for 24 h at 25 ± 0.2°C. The sorption experiments were carried out by adding 100 mL of P-containing solution in 0.1 M KCl at pH 5.0 to bottles with 100 mL of goethite suspension. The initial total P concentration was 522 µM for both IHP and Pi. After 30 h, 4 d, or 8 d of pre-sorption the suspensions were centrifuged. The supernatants were filtered through a 0.22-μm membrane and analyzed for P. The sediments were rinsed with 0.1 M KCl at pH 5.0 three times to remove the residual P and used for the desorption experiments.
The desorption kinetic experiments of IHP and Pi were initiated by adding 100 mL of H2O at pH 6.0, 0.02 M KCl at pH 6.0, or 0.02 M citrate at pH 6.0 to the sediments with pre-sorbed P. The bottles were shaken and at the selected time intervals (0.08, 0.17, 0.33, 0.5, 1, 3, 6, 12, 24, 48, 72, and 96 h) 5 mL suspension was withdrawn and immediately filtered through a 0.22-μm membrane. The filtrate was used for P analysis. To study the effect of pH on desorption in the presence of citrate, the desorption experiments were also performed at pH 4.0 and 8.0.
2.6 P analysis
The amount of Pi in the supernatants was measured with the phosphomolybdate blue colorimetric method (Murphy and Riley, 1962). Before the colorimetric determination, IHP was hydrolyzed to Pi by digestion with concentrated sulfuric and perchloric acids (Martin et al., 1999). The percentage recovery of this method was between 96.4 to 100.5% with a coefficient of variation less than 5% for organic P determination (Martin et al., 1999). Each P-concentration determination was conducted in triplicate and the average was used to calculate the sorption or desorption amounts. Losses of IHP by hydrolysis and sorption to the container walls were negligible.
2.7 ATR-FTIR spectroscopic analysis
ATR-FTIR spectra of IHP/Pi in solution and those sorbed on goethite were recorded on a FTIR spectrometer (Bruker Vertex 70, Ettlingen, Germany) equipped with a deuterated triglycine sulfate detector. A single-reflection diamond ATR accessory (Pike Technologies, Wisconsin, USA) was used to acquire the spectra of IHP and Pi solutions prepared at different pH (2–12). Additionally, IHP and Pi sorption onto goethite at pH 5 and 0.1 M KCl and the desorption by 0.02 M citrate at pH 6 were probed with an ATR-FTIR flow-cell setup similar to that described by Wang et al. (2013). Briefly, a thin and evenly coated goethite film was deposited onto a ZnSe crystal element in a horizontal 45° ATR cell (Pike Technologies, Wisconsin, USA). The film was prepared by placing a finely dispersed suspension of goethite (2 mg goethite in 1 mL of water) onto the crystal and drying overnight. The goethite deposit was first equilibrated with 0.1 M KCl at pH 5 for approx. 4 h. After this time, a background spectrum was collected as the average of 512 scans at a 4 cm−1 resolution in the spectral range of 1800−800 cm−1. Then the sorption experiment was started by pumping 60 μM IHP (or Pi) in 0.1 M KCl electrolyte at pH 5 into the reaction vessel at a flow rate of 0.5 mL min−1. At regular time intervals spectra were recorded and after 3 h of IHP (or Pi) injection, the final sorption spectrum was collected. Subsequently, 0.02 M citrate at pH 6 was pumped to the reaction vessel to desorb P from goethite. Sample spectra at different desorption times were collected; for each spectrum 512 scans were collected at 4 cm−1 resolution in the spectral range of 1800−900 cm−1.
2.8 Statistical analysis
All sorption and desorption isotherms were performed in triplicate. The data were presented as the means ± standard error of the mean (SEM). When not indicated, the SEMs were within the dimensions of the symbols shown on the graphs. The values of parameters for all sorption and desorption isotherms were fitted using Origin 8.0 (OriginLab Corporation, Northampton, MA, USA) at the 95% confidence level.
3 Results
3.1 Goethite characteristics
The XRD pattern of the synthesized goethite showed all appropriate peaks for this mineral and the absence of extraneous peaks confirmed its purity. The TEM image revealed acicular goethite crystals of approximately 1 μm in length and 40 nm in width; the specific surface area was 46.4 m2 g−1, within the range of values previously reported for goethite (45.0–55.0 m2 g−1) (Ruttenberg and Sulak, 2011; Johnson et al., 2012).
The ζ potential of goethite in 0.001 M KCl changed from positive (+53.3 mV) to negative values (–46.6 mV) when the pH increased from 3.0 to 10.0. The PZC in 0.001 M KCl was at pH 8.7 (data not shown). The ζ potential of pure goethite at pH 5.0 in 0.001 M KCl was stable over time at a value of about +40 mV (data not shown). After the addition of IHP, the ζ potential dramatically changed to –50 mV and then increased relatively fast to about –20 mV followed by a gradual increase in time to about –5 mV (300 h). After the addition of Pi, the ζ potential rapidly changed to about –5 mV, after which the potential further decreased slightly and then increased gradually to about +5 mV. Initially, the ζ potential strongly decreased upon the addition of IHP or Pi and reversed its sign, indicating that the positive surface charge of goethite was overcompensated by the negative charge of the sorbed P.
3.2 Sorption isotherms
Q = a · Qm · Ce / (1 + a · Ce),
()where Q is the equilibrium sorption density (µmol m−2), Qm corresponds to the pseudo-maximum sorption (µmol m−2), Ce is the equilibrium concentration of P in solution (µM), and a (L μmol−1) is the sorption affinity. The calculated Qm values of IHP and Pi on goethite were 0.62 and 1.98 μmol m−2, respectively. The calculated a, corresponding to the affinity of goethite for IHP (45.89 L μmol−1), was much higher than that of Pi (0.73 L μmol−1).
3.3 Desorption isotherms
The total desorption percentages of IHP and Pi from goethite after three desorption cycles are plotted in Fig. 1 as a function of the pre-sorption density for the three different desorbing reagents. The desorption amount increased with increasing sorption density, but much less than the sorption amount. Desorption by 0.02 M citrate was far more effective than that by 0.02 M KCl at pH 6. In water, the maximum desorption of IHP was low, but it was still more than twice that of 0.02 M KCl (7.3% by H2O vs. 3.5% by KCl). For Pi the maximum desorption was slightly higher in H2O than in 0.02 M KCl (23.0% by H2O vs. 18.3% by KCl). For IHP a maximum desorption of about 30% by citrate was observed at an initial sorption density of 0.65 μmol m−2, while for Pi, the maximum desorption was 60% at an initial sorption density of 2.2 μmol m−2.

Total desorption percentages of (a) myo-inositol hexakisphosphate (IHP) and (b) phosphate (Pi) from goethite by 0.02 M KCl, H2O, and 0.02 M citrate at pH 6.
The desorption curves of IHP (Fig. 2a, b) and Pi (data not shown) from goethite by the three different desorbing reagents are depicted in a different way, i.e., the desorption amount (expressed as solution concentration in μM) is plotted vs. the sorption density in μmol m−2. The desorption amounts in the first desorption cycle were the largest, accounting for > 50% of the total desorption, and decreased in each subsequent cycle.

Desorption curves of myo-inositol hexakisphosphate (IHP) from goethite by (a) 0.02 M KCl, (b) H2O, (c) and 0.02 M citrate at pH 6. Each data point represents the mean value from three repeats; in some cases, error bars are within the dimensions of the data points.
Cd = a · eb · Q,
()Cd = c + k Q,
()where c (μM) and k (m2 L−1) are constants. The equations for the first cycle and for the total desorption after three cycles are shown in Table 1 for all conditions.
| The first desorption cycle | r2 | The total of the three desorption cycles | r2 | |
|---|---|---|---|---|
| IHP | ||||
| KCl | Cd = 0.0045e10.37Q | 0.949 | Cd = 0.0098e9.35Q | 0.956 |
| H2O | Cd = 0.11e6.41Q | 0.993 | Cd = 0.33e5.36Q | 0.994 |
| Citrate | Cd = –5.68 + 49.01Q | 0.992 | Cd = –5.02 + 77.74Q | 0.994 |
| Pi | ||||
| KCl | Cd = 0.16e2.73Q | 0.976 | Cd = 0.51e2.45Q | 0.977 |
| H2O | Cd = 0.18e2.88Q | 0.975 | Cd = 0.27e2.84Q | 0.985 |
| Citrate | Cd = –62.17 + 120.84Q | 0.976 | Cd = –75.02 + 163.52Q | 0.981 |
3.4 Desorption kinetics
The initial sorption density after 4 d equilibration was 0.75 μmol m−2 for IHP and 2.69 μmol m−2 for Pi. There was a rapid desorption of IHP by KCl and H2O in the first hour followed by a slow one (Fig. 3a). For the desorption of Pi by KCl and H2O there was a re-sorption stage associated with the gradual decrease in desorption (Fig. 3b). In the entire desorption process, the desorption percentages of IHP and Pi by KCl were lower than by H2O. Desorption of IHP and Pi by citrate also started with an initially rapid desorption followed by a slower one (Fig. 3). The desorption of IHP and Pi in the presence of citrate was much greater as compared with in the presence of KCl or H2O. The initial desorption rates for IHP were much lower than those for Pi (data not shown).

Kinetics of (a) myo-inositol hexakisphosphate (IHP) and (b) phosphate (Pi) desorption from goethite after 4 d pre-sorption by 0.02 M KCl, H2O, and 0.02 M citrate at pH 6.
Kinetics of P desorption by 0.02 M citrate at different pH levels indicate that for both IHP and Pi the competition strength of citrate increased with decreasing pH (data not shown). As a result, the desorption rate increased with decreasing pH and at all pH values the rates for Pi were higher than those for IHP (data not shown).
The pre-sorption density at 8 d was 0.75 μmol m−2 for IHP and 3.94 μmol m−2 for Pi. Compared to those of Pi, the desorption percentages of IHP by KCl and H2O (less than 2% and 5%, respectively) were much lower under varying pre-sorption times (Fig. 4). The desorption of IHP slightly increased with increasing pre-sorption time. For IHP, the highest desorption rate and desorption percentage were observed at the longest pre-sorption time in both KCl and H2O (data not shown).

Kinetics of myo-inositol hexakisphosphate (IHP) desorption from goethite with different pre-sorption time by (a) KCl and (b) H2O.
However, a pronounced effect of pre-sorption time on Pi desorption was observed, which is different from the case of IHP desorption (Fig. 5). The longer the pre-sorption time, the lower were the desorption and the initial desorption rate (data not shown). This observation was similar for KCl and H2O. Furthermore, after 20 h the desorption diminished likely due to re-sorption. The re-sorption percentage of Pi after short-term pre-sorption (30 h) was greater than that after long-term pre-sorption (4 d). This is because the sorption density of P on goethite is relatively low with short-term pre-sorption, which results in the greater potential for P (re-)sorption over time (up to 80 h).

Kinetics of phosphate (Pi) desorption from goethite with different pre-sorption time by (a) KCl and (b) H2O.
3.5 ATR-FTIR spectroscopy
ATR-FTIR spectra of the IHP and Pi sorption kinetics at pH 5 in 0.1 M KCl and the desorption kinetics by citrate are depicted in Fig. 6. During the sorption, the intensities of IR sorption bands from IHP and Pi surface complexes increased over time, which is likely due to the increased IHP and Pi sorption on goethite surface (Fig. 6a, b). The IR sorption bands of IHP surface complexes on goethite at 1166 cm−1 [νas(P–O in ≡Fe–PO
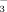 ) ], 1135 cm−1 [νas(P–O in PO32−)], 1075 cm−1 [νs(P–O in HPO
) ], 1135 cm−1 [νas(P–O in PO32−)], 1075 cm−1 [νs(P–O in HPO
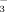 )], and 998 cm−1 [νs(P–O in PO32−)] show apparent differences from the IHP solution spectra at 1185 cm−1 [νas(P–O in HPO
)], and 998 cm−1 [νs(P–O in PO32−)] show apparent differences from the IHP solution spectra at 1185 cm−1 [νas(P–O in HPO
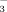 )], 1133 cm−1 [νas(P–O in PO32−)], and 1067 cm−1 [νs(P–O in HPO
)], 1133 cm−1 [νas(P–O in PO32−)], and 1067 cm−1 [νs(P–O in HPO
 )] (Fig. 6a). Similarly, the IR sorption bands of Pi surface complexes on goethite at 1096, 1050, and 951 cm−1 exhibit remarkable differences from the Pi solution bands at 1159, 1077, and 939 cm−1 (Fig. 6b). Generally, if the phosphate groups bind with metal ion(s) at the hydroxide surface by forming inner-sphere complexes, the bands corresponding to the vibration of phosphate groups will split into two or three bands or experience shifts (Guan et al., 2006). These shifts indicate that IHP and Pi were sorbed onto goethite by forming inner-sphere complexes. Similar shifts in band positions were observed in previous studies, in which the formation of inner-sphere complexes was proposed (Tejedor-Tejedor and Anderson, 1990; Guan et al., 2006; Luengo et al., 2006). Introduction of citrate promoted IHP and Pi desorption, as demonstrated by the decreasing intensities of IR vibrational bands of the P surface complexes (1200 to 900 cm−1) over time (Fig. 6c, d). Additionally, the intensities of IR sorption bands of citrate surface complexes at 1566 and 1392 cm−1 increased over time, reflecting an increasing citrate sorption on goethite surface with time. Also in this case, spectral changes occurred over the entire time span of the experiment, indicating that citrate sorption and P desorption progressed simultaneously.
)] (Fig. 6a). Similarly, the IR sorption bands of Pi surface complexes on goethite at 1096, 1050, and 951 cm−1 exhibit remarkable differences from the Pi solution bands at 1159, 1077, and 939 cm−1 (Fig. 6b). Generally, if the phosphate groups bind with metal ion(s) at the hydroxide surface by forming inner-sphere complexes, the bands corresponding to the vibration of phosphate groups will split into two or three bands or experience shifts (Guan et al., 2006). These shifts indicate that IHP and Pi were sorbed onto goethite by forming inner-sphere complexes. Similar shifts in band positions were observed in previous studies, in which the formation of inner-sphere complexes was proposed (Tejedor-Tejedor and Anderson, 1990; Guan et al., 2006; Luengo et al., 2006). Introduction of citrate promoted IHP and Pi desorption, as demonstrated by the decreasing intensities of IR vibrational bands of the P surface complexes (1200 to 900 cm−1) over time (Fig. 6c, d). Additionally, the intensities of IR sorption bands of citrate surface complexes at 1566 and 1392 cm−1 increased over time, reflecting an increasing citrate sorption on goethite surface with time. Also in this case, spectral changes occurred over the entire time span of the experiment, indicating that citrate sorption and P desorption progressed simultaneously.

ATR-FTIR spectra of (a) myo-inositol hexakisphosphate (IHP) and (b) phosphate (Pi) sorption kinetics on goethite at pH 5 and 0.1 M KCl, and (c) IHP and (d) Pi desorption kinetics by 0.02 M citrate at pH 6. The arrows indicate that the absorbance of the spectra increased or decreased with time.
4 Discussion
4.1 Comparison of sorption and desorption characteristics between IHP and Pi
On the basis of the ratio of sorbed IHP/Pi, it was proposed that the sorption of IHP most probably occurs via inner-sphere surface complexation with four phosphate groups in each IHP molecule being bound to goethite, while the other ones remaining free (Ognalaga et al., 1994; Celi et al., 2001). The tentative schematic conformation of IHP adsorbed on the surface of goethite is shown in Fig. 7a. However, the detailed structural configuration of the adsorbed IHP needs further investigation. The structural configuration of Pi on goethite is also proposed (Fig. 7b), which is in accordance with that of Kim et al. (2011). This may explain why the affinity of goethite for IHP (45.89 L μmol−1) is much greater than that for Pi (0.73 L μmol−1) and the maximum sorption density of IHP (0.62 μmol m−2) is two-thirds less than that of Pi (1.98 μmol m−2). The great affinity of IHP to goethite results in the higher irreversibility of IHP sorption and lower desorption percentages of IHP as compared with Pi (Figs. 3–5).

Tentative schematic conformation of (a) myo-inositol hexaphosphate (IHP) and (b) phosphate (Pi) adsorbed on the goethite (Gt) surface. For the sake of simplicity, the H atoms of the phosphate groups were omitted.
The initial decrease in the ζ potential of goethite sorbed with P is much larger for IHP than for Pi. This might be related to the fact that IHP is a large multivalent ion, which not only affects the charges within the plain of shear but also the position of the plain of shear. These results could also explain why the long-time potential adaptation processes are much more significant for IHP than for Pi. The increase in ζ potential later on might be due to the OH− release from the surface, which makes the ζ potential less negative and weakens the repulsion between the negatively charged IHP ions sorbed on goethite. It is also possible that some bound IHP ions protonate or form ternary surface complexes with cations (Fe3+), which may be followed by surface precipitation (transition from surface complexes to surface precipitates; Ler and Stanforth, 2003; Yan et al., 2014a). Similarly, at the initial stage, the sorption of phosphate enhances the negativity of the goethite surface, resulting in the strong decrease of ζ potential. The later gradual increase in ζ potential is probably due to the transformation of surface complexes to surface precipitates as proposed by Ler and Stanforth (2003). Such phenomena were also observed when IHP was sorbed on amorphous aluminum hydroxide and 5 nm γ-Al2O3 (Yan et al., 2014a, 2015).
4.2 Comparison of desorption mechanism between different reagents
The desorption percentages of IHP and Pi on goethite in the presence of citrate were much higher than those in KCl and H2O. This is mainly due to the fact that citrate also is adsorbed specifically on goethite (Lindegren et al., 2009) and, therefore, competes with P for sorption sites, as indicated by ATR-FTIR spectra (Fig. 6). The desorption amount and rate of IHP and Pi by citrate increase with decreasing pH, which may be related to the higher ability of citrate to compete with IHP when the pH is low (Celi et al., 2003; Lindegren et al., 2009), as reported for Pi (Geelhoed et al., 1998). However, Cl− and K+ are largely electrostatically bound in the diffuse part of the electrical double layer. In KCl or H2O, desorption is mainly driven by partition and diffusion processes. The removal of desorbed P occurs by diffusion from the region adjacent to the surface to the bulk solution (Koopal and Avena, 2001; Torn et al., 2005). The high affinity of P to goethite determines the high degree of irreversibility of the P sorption and thus low desorption. However, when P is removed from the surface by competition, the extra desorption reaction with ligands promotes desorption. In addition, the concentration gradient between the surface region and the bulk solution is sufficiently large to lead to substantial diffusional transport (Koopal and Avena, 2001; Torn et al., 2005). For KCl and water, the P gradient is very small and the transport by diffusion is also very small so that further desorption hardly occurs. At pH 6, goethite surface sorbed with IHP or Pi is negatively charged. The increase in ion strength makes the potential in the plane of adsorption less negative and thus increases adsorption, as indicated by Barrow et al. (1980). Therefore, the desorption of IHP/Pi by 0.02 M KCl is lower than that by H2O. The different desorption processes also lead to different desorption curves (Fig. 2) and equations. In the second desorption cycle the concentration of local P adjacent to the surface is lower than in the first cycle and, therefore, the diffusional transport becomes even lower, which explains less desorption (Koopal and Avena, 2001). In the third cycle, this effect is again stronger and further desorption is practically negligible.
4.3 Effect of pre-sorption time
Sorption of P on goethite involves two steps: a relatively fast step that takes place in the first few hours, and a slow step that takes days or weeks, and both processes can occur simultaneously (Luengo et al., 2006; 2007; Ruttenberg and Sulak, 2011). At short-term sorption, the reaction is mainly governed by the fast sorption stage during which P is sorbed to goethite surface by ligand exchange, and the reaction is assumed to be reversible, allowing for the release of sorbed P to the solution if environmental conditions favor desorption (McGechan and Lewis, 2002; Ruttenberg and Sulak, 2011). During the long-term sorption, however, the reaction is governed by the slow sorption stage during which P may diffuse into the interior of goethite aggregates or precipitate irreversibly on the surface, and both processes impede desorption (Froelich, 1988; McGechan and Lewis, 2002; Ler and Stanforth, 2003; Ruttenberg and Sulak, 2011). The presently observed initial desorption rates for Pi indeed show a slower rate at longer pre-sorption time and also a lower desorption (Fig. 5). However, for IHP the initial desorption rate increases at a long pre-sorption time and the desorption increases with pre-sorption time (Fig. 4). Although the large molecule of IHP can be one reason for its indistinctive diffusion into goethite in the long-term sorption, the present results of enhanced desorption with pre-sorption time for IHP cannot be fully explained by the above concept of two binding processes. The ζ potential results over time already indicate that the sorption of IHP as a function of time is a complex process, which deserves further investigation.
5 Conclusions
Desorption of IHP and Pi from goethite is affected by pre-sorption density, types of desorbing reagents, desorption cycles, pH, and pre-sorption time. In general, the desorption percentage and rate of IHP from goethite are lower than those of Pi. Desorption percentages and rates of IHP/Pi in citrate solution are much higher than those in KCl and H2O, and both parameters generally increase with increasing pre-sorption density. The desorption potential of IHP/Pi by citrate increases with decreasing pH. In KCl and H2O the desorption is mainly caused by sorption–desorption equilibrium and diffusion transport. The desorption of IHP/Pi in 0.02 M KCl is lower than that in H2O. An exponential equation fits well to the desorption curves of IHP and Pi from goethite in KCl or H2O, while a linear equation is more appropriate for citrate system. Low-molecular weight organic acids, such as citrate, can substantially enhance the mobility and availability of IHP/Pi originally sorbed on goethite surface via ligand exchange. This study contributes to the further understanding of the desorption behavior of P in the environment.
Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (Grant Nos. 41171197 and 41471194) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB15020402) for financial support of this research.

