

In silico study of SARS-CoV-2 spike protein RBD and human ACE-2 affinity dynamics across variants and Omicron subvariants
Abstract
The coronavirus disease 2019 virus outbreak continues worldwide, with many variants emerging, some of which are considered variants of concern (VOCs). The WHO designated Omicron as a VOC and assigned it under variant B.1.1.529. Here, we used computational studies to examine the VOCs, including Omicron subvariants, and one variant of interest. Here we found that the binding affinity of human receptor angiotensin-converting enzyme 2 (hACE2) and receptor-binding domain (RBDs) increased in the order of wild type (Wuhan-strain) < Beta < Alpha < OmicronBA.5 < Gamma < Delta < Omicron BA.2.75 < BA.1 < BA.3 < BA.2. Interactions between docked complexes revealed that the RBD residue positions like 452, 478, 493, 498, 501, and 505 are crucial in creating strong interactions with hACE2. Omicron BA.2 shows the highest binding capacity to the hACE2 receptor among all the mutant complexes. The BA.5's L452R, F486V, and T478K mutation significantly impact the interaction network in the BA.5 RBD-hACE2 interface. Here for the first time, we report the His505, an active residue on the RBD forming a salt bridge in the BA.2, leading to increased mutation stability. When the active RBD residues are mutated, binding affinity and intermolecular interactions increase across all mutant complexes. By examining the differences in different variants, this study may provide a solid foundation for structure-based drug design for newly emerging variants.
1 INTRODUCTION
Since 2019, the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has spread rapidly worldwide. It was declared a global pandemic and continued to gain interest due to its profound negative impact on health and the economy. However, the pandemic is yet to end,1 with more than 635 million confirmed cases and 6.4 million deaths reported by 2022. Since the first SARS-CoV-2 strain emerged in Wuhan, China, the virus has evolved, giving rise to several lineages and variants. The most common SARS-CoV-2 variant in the worldwide epidemic now carries the amino acid mutation D614G in the SP (spike protein). The dynamic tracking of variant frequencies discovered a recurring pattern of G614 rise across different geographical levels. The virus consists of several genomes combined with a single RNA strand and undergoes rapid mutations resulting in a high human transmission.2
The SP found on the surface of the virion is a type I fusion glycoprotein containing varieties of trimers with two subunits: S1 and S2. The S1 subunit on the viral membrane includes the N-terminal domain (residues 14−305), forms receptor-binding domain (RBD) residues 319−541 and is essential for replication.3 The RBD of the SP identifies the human angiotensin-converting enzyme receptor (hACE2R) in the respiratory system to enter the human host cell. The binding capacity of the SP of SARS-CoV-2 that emerged in Wuhan city (2019) displayed 10 to 20 times more affinity toward hACE2R than the virus that occurred in 2002.4
The WHO introduced the term “Variant of Concern” (VOC) to recognize the viral variants with mutations in their SP, which changed the binding affinity to the hACE2R.5 The VOCs include Alpha (B.1.1.7-lineage), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and, more recently, the Omicron (B.1.1.529) emerged in South Africa. Recent literature reports that the RBD of Omicron (B.1.1.529) acquired a strong affinity and binding capacity to hACE2R, resulting in higher transmissibility.6 Recent studies claimed that the Omicron is more highly transmissible than the previous variants with 16 mutations on the RBD.7 Therefore, it is essential to study the novel mutations in the Omicron-RBD variant to determine their interaction with the hACE2R.
The Omicron subvariants (OSVs) BA.1, BA.1.1, BA.2, and BA.5, which emerged in South Africa in late 2021, spread rapidly across Europe, Canada, Australia, and the USA, indicating their boosted virulency. As of July 2022, the Omicron BA.5 variant spread like wildfire throughout Japan. Similarly, first discovered in India, the Omicron BA.2.75 initiated an impending epidemic worldwide. It remains a variant of interest (VOI) and is under monitoring. Therefore, here, we aimed to conduct detailed modelling of the SP-RBDs of the VOCs, one variant of interest and the hACE2R using in-silico tools. Further, we seek to justify the current clinical setting observed of the Omicron variant and OSVs, including BA.2, BA.2.75, and BA.5.
2 METHODS
2.1 Protein data acquisition
The FASTA sequence of the SARS-CoV-2 spike glycoprotein of Wuhan-Hu-1 (wild-type [WT]) was obtained from the UniProt protein database (Accession no: P0DTC2). Amino acid substitutions of SP-RBDs were obtained from the global report platform and GSAID.8 The residue changes reported for VOCs and OSVs were manually incorporated into this sequence to obtain mutated SP-RBDs. The X-ray crystal structure 2.45 Å resolution complex of the WT SARS-CoV-2-RBD with hACE2 (PDB ID 6M0J) was retrieved from the Protein Data Bank to obtain the 3D structure of hACE2R.
2.2 In silico homology modeling and mutagenesis analysis
To generate the RBD variants, suitable substitutions of amino acids were made using the PyMol Mutagenesis plugin.9 The generated sequences were submitted to the SWISS-MODEL bioinformatics program to model the Protein 3D structures of different variants.10 The resulting structure was constructed by PyMol software by adding missing residues. The stability of each selected mutation was studied using the DUET-webserver11 to evaluate docking scores of predicted RBD structures.
2.3 Structure validation and refinement of variant structures
The quality of each 3D model of RBD and hACE2R was validated by two webservers, the structure analysis verification Server (SAVES 6.0)12 and the protein structure analysis (Pro-SA) webserver.13 The SAVES utilizes the Ramachandran plot to evaluate the models, and Pro-SA relies on Z-score or the overall model quality. The mutated RBD and hACE2R protein structures were refined using the Galaxy Refine webserver,14 and energy was minimized using the Chiron webserver 15
2.4 Protein−protein docking of mutated RBD and hACE2
Docking between hACE2 and RBD of different variants, OSV and VOI was performed using the “EASY” access level of the HADDOCK 2.4 server.16 The selected cluster models were downloaded in PDB format for further analysis. This was followed by selecting the first model with the lowest binding free energy value. Active/passive residues are introduced to provide information about the interacting residues and can be employed as restraints during docking. Based on intermolecular interactions within a 6.5 Å cut-off, the HADDOCK docking methodology establishes the protein−protein interface as flexible. Therefore, separate flexible zones may be established for each binding stance. An examination of intermolecular interactions automatically defines semiflexible residues.17
2.5 Determination of dissociation constant (KD) and comparison of interactions between mutant RBD and hACE2 receptor
The was conducted using the PROtein Binding Energy Prediction (PRODIGY), an automated server to calculate the binding affinity and values for different biological complexes. The binding affinities were calculated using PRODIGY.18 The interaction profile of spike RBD-hACE2R complexes was further explored through PDBsum19 and PyMol 2.5.2.9
2.6 Molecular dynamics (MD) simulations
Classical MD simulations were performed using GROMACS 2020.6 software to assess the complexes' structural stability and flexibility.20 The 20 ns simulation was carried out for the RBD mutant complexes of the WT, delta, BA.2, and BA.5. Protein complexes were relaxed with an AMBER99SB-ILDN force field and defined a cubic box around it with a 2.5 nm distance from the box edges.21 A protein complex was added to a TIP3P water box, solvated, and neutralized by adding counter ions. MD simulations were carried out for a timescale of 20 ns using the protocol of Khan et al.22 Subsequently, the root means square deviation (RMSD) for each protein backbone was estimated using the actual structures of each protein as a guide. After 20 ns, the MD trajectories were analyzed by obtaining the complex's RMSD plot.23
2.7 Data analysis and visualization
Interaction analysis graphs, line graphs, and bar charts were plotted using GraphPad Prism-9.4.024 and Origin 2022b for windows. All 3D representations were done using VMD 1.9.3 or PyMol 2.5.2.
3 RESULTS
3.1 Mutational changes in RBD SPs and structure stability
The enhanced virus infectivity may result from an improved capacity to bind to the host receptor with more excellent stability. Thus, we examined each variant's strength of each 21 RBD mutation (Table 1).
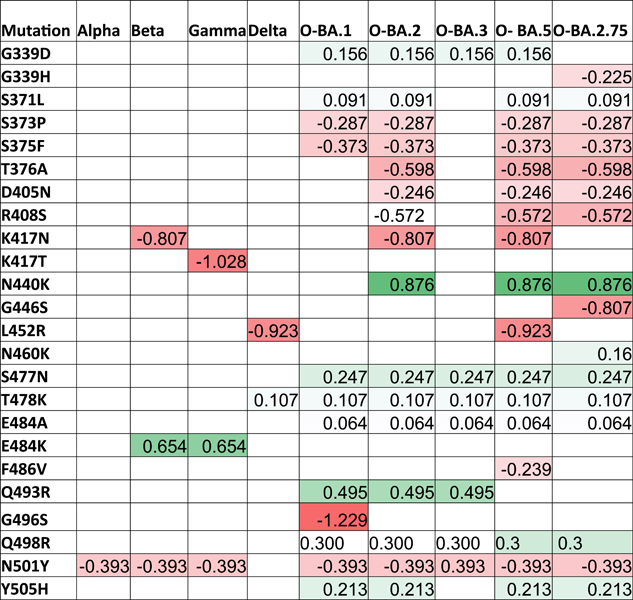 |
- Note: Red color: destabilizing mutations; Green color: stabilizing mutations. All the stability values are presented in kcal/mol.
- Abbreviations: D, decreased stability; De, destabilizing; In, increased stability; O, Omicron; RBD, receptor-binding domain; S, stabilizing; VOCs, variants of concern.
Missense mutations in RBDs could affect the binding affinity to hACE2R. We used the DUET webserver to evaluate missense mutations in the RBD protein and to assess the protein stability associated with mutations. The stability value (ΔΔG kcal/mol) for each mutation altered the overall stability of each VOC and OSV. The mean ΔΔG values in monomer stability at each full-length SP-residue position ranged from −1.229 kcal/mol in G496S to 0.876 kcal/mol in N440K. We illustrated the AA positions that firmly stabilized or destabilized the SP in Figure 1 and listed their percentages for each VOC and subvariant in Table 1. The G339D, S371L, N440K, S477N, T478K, E484A, E484K, Q493R, Q498R, and Y505H mutations in the glycoprotein residues showed maximum stabilizing effects on the full-length SP. Also, only 47.6% of the 22 unique AA substitutes exhibited stabilizing effects, while 52.3% of mutations showed destabilizing effects on the SP. Notably, the N440K (ΔΔG: 0.876 kcal/mol) transformation that occurred in BA.2 and BA.5 exerted a high stabilizing effect on the RBD region with a high ΔΔG value. Comparing the mutations that took place in the RBDs of Beta and Gamma (E484K; ΔΔG: 0.654 kcal/mol) and Q483R (ΔΔG: 0.495 kcal/mol) in OSVs, Beta and Gamma exhibited a highly stabilizing effect. However, mutations in Delta and BA.5 (L452R) and Beta, BA.2, and BA. 5 (K417N) exerted a higher destabilizing impact on the RBD region with low ΔΔG values, which predicting the effects of missense mutations on protein stability (Figure 1).

3.2 Homology modeling for mutagenesis analysis and structural validation
We created 3D-RBD protein structures for each variant using the SWISS-MODEL server.10 For further examination, the models that best matched the viral spike RBD were chosen and compared with the WT. The Alpha variant RBD contains one AA substitution (N501Y), while Beta and Gamma acquired three compared to the WT. On the contrary, Delta exhibited two novel substitutes (L452R, T478K). The N501Y substitution was seen in Alpha, Beta, and Gamma and BA.1 and BA.2 and the E484K mutation exists in Beta and Gamma.
Except for the Delta, all VOCs included the substitution N501Y. The N501Y and T478K were notable alterations in the Omicron-variant's RBD region. The T478K substitution was previously found in the Delta. The BA.5 showed L452R and F486V substitutions in its RBD motif. Among the mutations, L452R was once prominent only in the Delta. The BA.5 mutations overlapped with BA.2 except for the Q493R modification.
Further validation of the homology modeling protein structures was performed using ProSA-Web. The overall model quality of the RBD models of the WT, VOCs, and OSV is presented in Supporting Information: Table S1. The Z-score ranges from −0.51 to –1.81. Since all Z-score values are negative, the modeled structures appeared valid. We used the Ramachandran plot to validate the overall quality of stereochemistry. The plot resulted in four areas: most favored, additionally allowed, generally allowed, and disallowed (Supporting Information: Figure S1). The modeled RBD structures of the WT and mutants exhibited >90% AA residues in the most favored region and zero non-glycine residues in the disallowed regions, thus indicating the high quality of structures.
3.3 Comparative analysis of the binding capacity of mutant RBD complexes to hACE2
The mechanisms of the binding interactions and the affinities between RBDs and hACE2 of the WT and the variants were elucidated using the HDOCK server. The results of the docking scores, cluster size, and KD are given in Table 2, and the docking complexes in Supporting Information: Figure S2. The predicted HADDOCK score for the WT-hACE2 complex was −87.7 kcal/mol, the lowest binding affinity among the studied complexes. Comparing WT, Delta, and Omicron variant's decreased docking score indicated higher binding affinity. Among OSVs, prediction for docking score with hACE2 was seen in the BA.2 with a 1.5-fold increased affinity (−130.1 kcal/mol). However, the score did not vary significantly among other OSVs (BA.1: −102.8; BA.5: −95.6 kcal/mol), comparable to the Delta (−99.2 kcal/mol). The newest AA substitutions in the BA.5 displayed a 1.3-fold less affinity than the BA.2 and a onefold increased binding capacity than the WT. These results emphasize the dynamic of binding affinity of each variant to the hACE2R.
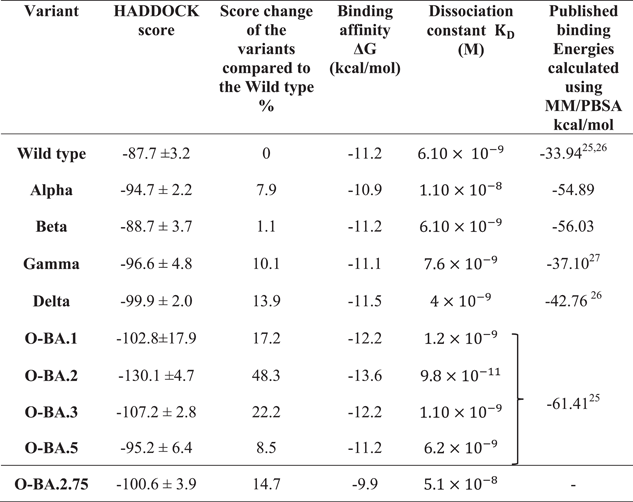 |
- Note: Binding affinity and dissociation constant were generated from PRODIGY web server.
- Abbreviations: hACE2, human receptor angiotensin-converting enzyme 2; O, Omricon; OSVs, Omicron subvariants; RBD, receptor-binding domain; VOCs, variants of concern; WT, wild-type.
Similar trends were observed with binding affinity values. The BA.2 predicted an affinity value of −13.6 kcal/mol, compared to the WT exhibiting an increase (−11.2 kcal/mol) in binding capacity. Compared to BA.2, other OSVs showed (BA.1 and BA.3 and BA.5) lower binding affinities. We also determined the KD and analyzed the changes in RBD-hACE2 complexes upon binding. The obtained KD values followed the same patterns of docking scores and prodigy predictions. The highest complex stability (160-fold) was evident in the BA.2-hACE2 complex (KD: 9.8 × 10−11 M) compared to the WT complex (KD: 6.1 × 10−9 M). The binding stability of hACE2 and RBD complexes increased in the order of BA.2.75 < Alpha < BA.3 < BA.1 < Delta < WT = Beta < BA.5 < BA.2 due to the decrease of the KD.
To evaluate the binding variations between hACE2 binding with RBDs of VOCs, subvariants, and the WT, we studied the RBD mutation interaction categories and numbers in detail. The nonbonding interactions increased in the order of WT < Beta = Delta < BA. 4/5< Alpha < BA.1 = BA.3 < Gamma < BA.2 with the hACE2R (Supporting Information: Figure S3).
The receptor interaction study showed salt bridge formation between Glu38, Glu39, and Lys323 of WT-RBD, with Lys417, Arg403, and Glu484 of the hACE2, respectively. Additionally, the WT revealed 10 hydrogen bonds and 71 nonbonded interactions (Figure 2I). In the Alpha complex, the N501Y substitution destroyed the salt bridges seen in the WT (Figure 2II), forming new favorable nonbonded interactions with Tyr449 and Gly496. Besides, the number of nonbonded interactions increased in the Alpha compared to the WT, which may have led to the increased binding capacity of the Alpha complex.





The Delta complex displayed interactions between 17 RBD and 20 hACE-2 residues, while the BA.2 displayed interactions between 26 RBD and 24 ACE2 residues (Figure 2V,VII). The new salt bridge found in the Arg408 of the Beta complex (Figure 2III) was absent in WT and Alpha. The number of nonbonded interactions in the Beta is lower than that of the Alpha and slightly higher than the WT (Supporting Information: Figure S3). Docking scores of the WT, Alpha and Beta are proportional to the number of nonbonding interactions. The residue Arg403 forms a salt bridge in WT and Gamma. While Arg 403, Gln 498, Asn 501, and Gln 493 generated hydrogen bonds in WT and Gamma. However, only the Gamma Tyr 505 residue formed H-bonding (Figures 2I and IV). However, compared to the WT with 71 nonbonded interactions, the Gamma exhibited only 124 interactions (Figure 2IV). This could explain the increased binding capacity of the Gamma compared to the WT.
The AA alteration in the BA.2 changed the WT-RBD Tyr505 residue into His505 (Figure 2VIIb) and was absent in other variants (Figure 2VI−IX). The Tyr505 residue in the WT was essential for nonbonded interaction, and interestingly the substituted AA in the BA.2 also formed an H-bond interaction. The Gln493 AA residue helped to create connections in all mutant complexes. But in the Omicron variant, Glnb493 residue converted to Asn493 but was absent in the Asn493 residues of the BA.5. (Figure 2IX), which produced H-bond interactions with the hACE2 in WT, Alpha, Beta, Gamma, BA.1 and BA.3 (Figure 2I−IX c). The only BA.2 and BA.5 complexes did not generate H-bonds in Asn493 RBD residues (Figure 2VI,IX). Except for this Asn493, all other substitutions found in the BA.1 were also seen in BA.5. However, the Arg452 mutation, seen only in the Delta, reappeared in BA.5. In BA.1 and BA.3, the residue Arg498 participated in the formation of a salt bridge and two H-bonds (Figure 2VI c). Yet the H-bonds found in the Arg498 of the BA.2 were absent in the BA.5, which may have led to lowering the binding affinity of the BA.5 compared to Delta and the OSVs. The Arg498 exhibited the highest DUET projected stability, indicating a high stabilizing mutation. Same as BA.2, BA.2.75 also forms a salt bridge between the His505 residue of RBD and Glu38 of hACE2R.
Different types of interaction between spike RBD and hACE2R can be identified as polar−polar, charged−apolar, polar−apolar, apolar−apolar, and charged−charged interactions (Supporting Information: Table S2). DIMPLOT figures show the hydrogen bonds between each mutant complex (Figure 3).



3.4 MD simulations
An MD simulation was conducted to analyze the binding stability of hACE2-RBD complexes, where multiple descriptors were interpreted to understand the flexibility and stability of the complexes. We employed RMSD as a time function and computed each complex's stability. The RMSD analysis of WT-RBD, Delta (Figure 4a-light green), and BA.2 (Figure 4a-light blue) revealed minor oscillations, indicating stabilized complexes. The BA.5 showed significant fluctuations (Figure 4a-dark blue), suggesting that it is yet to be stabilized. The WT complex stabilized after 2 ns, while the Delta offers a 6.5 Å divergence between 0 and 6 ns. However, after 6 ns, a relatively minor fluctuation was observed in the RMSD plot. In the BA.2 complex, the RMSD value was reduced to 3 Å and retained for the rest of the simulation. The RMSD of the BA.5 did not converge substantially but increased gradually. During the first 0−11 ns, the RMSD remained higher at 8.0 Å, and between 12 and 20 ns, the RMSD decreased to 4.5 Å.

The radius of gyration (Rg) can be used as an indicator of the compactness of the protein structure during the simulation period. All the facilities met increased and declined Rg values throughout the simulation. The average Rg values for WT, Delta, BA.2, and BA.5 were 33, 34.5, 36, and 34.5 Å, respectively (Figure 4b), with Rg values of BA.2 and BA.5 showing considerable fluctuation. Usually, the binding and unbinding of one or other end of the spike RBD domain trigger changes in the Rg value at various intervals in all the system.25 Since the MD simulation was limited to 20 ns, we could not draw a solid conclusion on each molecule's behavior. In the case of the Delta, the Rg value remained comparatively higher.
4 DISCUSSIONS
SARS-CoV-2 is still rapidly evolving, resulting in genetic heterogenicity in the viral population. The hACE-2 facilitates the entry of the virus on the cell surface, which binds to the RBD of the SP. The mutations in the SP could enhance the viral entry into the host cell, and the RBD could affect viral infectivity and stability.26 The development of novel drugs primarily focuses on the interaction interface.27 A mutation at a particular site or interface may directly impact binding and expression. Therefore, identifying specific protein binding aids in developing novel therapeutic strategies, particularly considering the relationship between the spike glycoprotein's RBD and the hACE2R. Therefore, this study examined the effects of RBD mutations on the binding affinity and stability across different VOCs from the WT to the BA.5 and BA.2.75.
The absolute quality of the model is created in terms of how well the model scores agree with the expected values of a high-resolution experimental structure. The QMEAN scores fell within −4.0 and near zero, demonstrating the high quality of the RBD structures.28 When the percentage of AA residues in the preferred region was >90%,29 and the percentage of non-glycine residues in the disallowed region was <15%,30 the protein structure was said to be of high quality. The modeled structures in this study matched the requirements described by Lovell et al.29, 30
Recent variants reported in the United Kingdom (Alpha- B.1.1.7), South Africa (Beta- B.1.351), and Brazil have shown significant structural deviation and changes in binding properties.31 We observed one mutation in the RBD (N501Y) in the Alpha compared to the WT. We discovered that the Alpha had a 7.9% binding capacity than WT, which could have led to increased coronavirus cases reported with the Alpha compared to the WT.32 The Lys478 residue formed a salt bridge and one hydrogen bond in the Delta complex and was absent in the Alpha, Beta, and Gamma complexes. However, the same residue reappeared in the BA.5, forming strong interactions. This might indicate a connection between the mutation's relationship and the variant's transmissibility.6 We observed the formation of a salt bridge between K417 (RBD) and D38 (hACE-2) in the WT RBD-hACE2 complex confirming the results obtained by Wang et al.33 However, the K417 was substituted by N417 and T417, respectively, in Beta and Gamma, resulting in the loss of the salt bridge. According to Han et al.34 and Li et al.,35 the absence of salt bridges reduced the binding affinity for hACE-2. However, our results contradicted their observations. We obtained high binding energy for the Gamma compared to the WT, even without a salt bridge formation, highlighting the importance of other intermolecular interactions for increased binding capacities.
Binding affinity (- kcal/mol) defines whether any complex formation occurs or not and holds a vital role in controlling interactions. Except for Delta and Omicron, other VOCs showed higher KD and weaker binding affinity values, confirming the results of Khan et al.31 In this study, the BA.2 exhibited the lowest binding affinity (−13.6 kcal/mol), proving the high stability of the complex compared to other variants. The BA.2 showed the lowest KD value (9.8 × 10−11) and high hACE-2 affinity compared to other VOCs and OSVs. When the KD value is reduced, the SP-RBD binds to hACE-2 strongly, creating a greater affinity between receptor and ligand.36
In the present study, BA.2 significantly alters the binding affinity with changes in bonding patterns compared to other VOCs. Six crucial AA residues, Leu455, Phe486, Gln493, Ser494, Asn501, and Tyr505 in RBDs, are essential for strong interaction in RBD-hACE-2 complexes.37 Further, they confirmed that Lys31, Glu35, Asp38, Met82, and Lys353 residues in the hACE2R are critical to making tight interactions at the active site. Among the mutations, Alpha, Beta, Gamma, and Omicron variants contained N501Y mutation in their RBDs, which is a primary concern since it was one of the active residues that directly interact with the hACE2R.6
The HADDOCK scores indicated that the Delta (−99 kcal/mol) and the OSVs have more excellent binding abilities than the WT (−87kcal/mol). The RBD of the Omicron (B.1.1.529) has undergone considerable mutation. The BA.1 and BA.2 became more prevalent since they initially appeared and are the most pervasive OSVs. Among the subvariants, the lowest score (−130.1 kcal/mol) was observed in BA.2, which agrees with Han et al.38 The data presented in this report justifies the higher transmissibility seen in the Omicron variant and subvariants.39 Interesting research suggests that the BA.2 Omicron subvariant is 1.5 times more contagious than BA.1 according to an in vitro experiment employing human nasal epithelial cells40 and it confirmed this highest binding energy of that variant. Omicron BA.1 was predicted by a recent study to be 10 times and 2.8 times more contagious than Wuhan-Hu-1 and Delta variants, respectively, primarily because of its RBD mutations N440K, T478K, and N501Y.41 Omicron's overall infectivity is far higher than the ancestral SARS-CoV-2 variation and other later variants, such as Delta, primarily because of its enormous number of mutations in the RBD regions.42
According to the previous findings, AA substitutions, Q493K and Q498H improved the RBD's affinity for binding with hACE2.43 In our study, the AA substitution occurred in the 498th position of the RBD involved in forming salt bridges and hydrogen bonds in the BA.1 and BA.3. The BA.3 formed a salt bridge between Arg498-Glu38 as confirmed by Ali and Vijayan.44 However, BA.2 formed hydrogen bonds at the 498th position. This may have enhanced the complex stability and hACE2R attachment. In the BA.5, the 493rd residue did not include any significant interaction, resulting in an increased docking score or lower binding affinity than OSVs.
The docking scores of the Delta and the Alpha are like that of the BA.5. It may be due to AA substitutions at L452R and T478K. The above mutations and F486V have not been observed in the BA.2. Like the Delta, the 478th residue at RBD of the BA.5 formed a salt bridge and hydrogen bond with the hACE2R. The same Delta, Arg452 mutation was observed in the BA.5, where the residue was involved in forming two hydrogen bonds and one salt bridge, thus increasing the binding affinity compared to Delta. Although the BA.5 had a lower binding affinity than the BA.2 and showed enhanced pathogenicity and efficient spreading in the human,45 suggesting that other factors are also involved in the viral transmission other than the binding energy.
We found that the His505 mutated in all OSVs but only in the BA.2; His505 is involved in developing a salt bridge. Also, the His505 is an active residue on the RBD, and this is the first report on the active residue involved in forming a salt bridge. This was further confirmed by DUET server results, where the mutation from Tyrosine to Histidine in 505 positions led to increased mutation stability. The results contradicted the study by Han et al.,38 where they observed that the His505 established fewer interactions with hACE2R. They justified that the lower binding affinities obtained for Omicron-RBDs were due to the Y505H.
When considering the active site residues of RBDs, the 493 showed hydrogen bond formations in OSVs complexes except in BA.2 and BA.5. Hydrogen bonding is crucial for docking score value.
When comparing BA.1, 2, and 5, the BA.5 resists neutralization by triple-dosed vaccinee serum more than BA.1/2, which can be attributed to L452R and F486V mutations in the BA.5 that contribute toward escaping of monoclonal antibodies.46 Further docking analysis is needed to prove the antibody escape by analyzing the attachment of RBD of these subvariants with monoclonal antibodies.
The deviations observed during the simulation time were used to evaluate how a biological molecule changed from its initial conformation. More minute fluctuations imply that the structure is stable and only exhibits the vibrations that occur naturally.6 We selected WT, Delta, BA.2, and BA.5 for RMSD analyses based on their docking scores. The average RMSD values for the WT, Delta, BA.2, and BA.5 were 3.5, 3.0, 3.0, and 4.5 Å, respectively, suggesting relatively stable complexes. However, the RMSD of the WT system remained uniform, with significant perturbation observed between 16 and 18 ns. However, extended simulation is required to understand the dynamic behavior deeply. When comparing BA.2 and BA.5 to the WT and Delta, high RMSD fluctuations were observed, suggesting that the WT and Delta RBD were more stabilized than BA.2 and 5. Ranjan et al.47 obtained similar changes with OSVs. The WT Rg value appeared to be more compact than Delta, BA.2, and BA.5, possibly due to the binding and unbinding at either end of the spike RBD domain.31
5 CONCLUSION
Five VOCs, the WT, and four OSVs were used to examine conformational changes and their impact on protein stability. The increased number and quality of interactions between the Omicron SP-RBDs and the hACE2R suggested increased potency. This study provides a biophysical basis for the high transmissibility of OSVs. Docking scores and interaction of mutated residues of the BA.5 are like that of Delta. When the active RBD residues are mutated, we observed increased binding affinity and intermolecular interactions across mutant complexes. The data presented in this report justifies the higher transmissibility seen in the Omicron variant, especially in the BA.2 variant. Finally, by examining the intermolecular interactions in different variant complexes, this study may provide a solid foundation for structure-based drug design for OSVs.
AUTHOR CONTRIBUTIONS
Conceptualization: D. C. P. Data collection and methodology: S. A., M. P., and D. C. P. Software, validation, and formal analysis: S. A. Data curation: S. A., U. B., and M. P. Analyzed the data and wrote the manuscript: S. A. and U. B. Reviewing and editing: D. C. P., U. B., D. P., and M. P. Visualization: S. A., U. B., and D. P. Supervision: D. C. P. Project administration: D. C. P.
ACKNOWLEDGMENTS
We are grateful to Mr. K. Sellahewa, Department of Physics, University of Sri Jayewardenepura, for his assistance in improving figure quality and Mr. D. P. P. Meddage for revising the manuscript with edited figures. The study was funded by the University of Sri Jayewardenepura grant (ASP/RE/SCI/2022/13).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Open Research
DATA AVAILABILITY STATEMENT
The data generated and analyzed during the current study are available from the corresponding author upon request.


{kind=link}