

Spin states in polynuclear clusters: The [Fe2O2] core of the methane monooxygenase active site
Abstract
The ability to provide a correct description of different spin states of mono- and polynuclear transition metal complexes is essential for a detailed investigation of reactions that are catalyzed by such complexes. We study the energetics of different total and local spin states of a dinuclear oxygen-bridged iron(IV) model for the intermediate Q of the hydroxylase component of methane monooxygenase by means of spin-unrestricted Kohn–Sham density functional theory. Because it is known that the spin state total energies depend systematically on the density functional, and that this dependence is intimately connected to the exact exchange admixture of present-day hybdrid functionals, we compare total energies, local and total spin values, and Heisenberg coupling constants calculated with the established functionals BP86 and B3LYP as well as with a modified B3LYP version with an exact exchange admixture ranging from 0 to 24%. It is found that exact exchange enhances local spin polarization. As the exact exchange admixture increases, the high-spin state is energetically favored, although the Broken-Symmetry state always is the ground state. Instead of the strict linear variation of the energy splittings observed for mononuclear complexes, a slightly nonlinear dependence is found. The Heisenberg coupling constants JFe1Fe2—evaluated according to three different proposals from the literature—are found to vary from −129 to −494cm−1 accordingly. The experimental finding that intermediate Q has an antiferromagnetic ground state is thus confirmed. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 1223–1239, 2006
Introduction
Polynuclear transition metal complexes play an important role as catalysts not only in technical and synthetical applications, but also serve as essential building blocks in the active centers of various enzymes.1-3 Prominent examples of transition metal containing sites in enzymes that are essential for life on earth are the iron–molybdenum cofactor (FeMoco) of nitrogenase,4-6 which catalyzes the conversion of atmospheric dinitrogen to ammonia in nitrogen-fixating bacteria, the Mn4Ca site within photosystem II,7 where the photosynthetic oxidation of water takes place, and the diiron center in the hydroxylase component (MMOH) of methane monooxygenase (MMO) found in methanotropic bacteria that convert methane to methanol.8, 9
To understand their functionality, the geometric and the electronic structure of these enzymes, in particular of their active sites, needs to be known. The experimental determination of the geometric structure is possible via X-ray crystallography. However, in cases where a protein cannot be crystallized, where the state under study is too labile or too short-lived, or where for other reasons the spatial and/or time resolution is not sufficient, X-ray crystallography cannot provide a detailed picture of the active site structures, so one has to rely on information from other spectroscopic techniques such as extended X-ray absorption fine structure (EXAFS) or EPR/ENDOR. Further information on the electronic structure can be extracted from temperature-dependent magnetic susceptibility measurements,10 which yield qualitative and quantitative information on the energetics of different spin states expressed by the sign and the magnitude of the Heisenberg coupling constants J.
It is not always possible to deduce without ambiguity from the sum of all avaliable spectroscopic data which geometric structures are present in the active site of an enzyme during reaction. Quantum chemical calculations can help to distinguish between different suggested structures by identifying the one that agrees best with all experimental data. For one recent example of EPR calculations on an oxygen-bridged manganese complex that may serve as a Mn4 photosystem II model, see ref.11. Once confidence in the theoretical methods is established by such comparisons, theoretical studies may be used in a predictive way for tackling mechanistic aspects.
Several factors affect the accuracy with which quantum chemical calculations reproduce or predict experimental results. First, one either has to build a truncated model of the active site or restrict the quantum chemical description of the system under study to a limited region within an embedding scheme like a QM/MM treatment (see, e.g., ref.12), because a complete enzyme is not accessible by current first principles methods. Second, the results are affected by the ability of the chosen quantum chemical method to describe the complicated electronic structure of transition metal complexes correctly. At present, the only method that is suited for the treatment of such systems of a reasonable size is density functional theory (DFT). Within DFT, the choice of the basis set and of the approximate exchange–correlation functional as well as the degree of control over the state to which the self-consistent-field (SCF) algorithm converges have considerable influence onto the results. Because it is straightforward to improve the basis set systematically, the focus of this study is on the effect of the exchange–correlation functional on various molecular quantities such as total energies, Heisenberg coupling constants, and local spins, as well as on the stability of SCF convergence. Because systematic investigations are very rare, we aim at a detailed account that concentrates on the essential parameters.
The identification of essential parameters for such a study is guided by the observation that the absolute and the relative total energies of various mononuclear transition metal complexes in different spin states depend linearly on the exact exchange admixture,13 that is, on the admixture of Hartree–Fock-type exchange interaction. It is the exact exchange contribution in present-day density functionals that makes the difference rather than a specific set of optimized parameters or functional contributions. A reparametrization of the exact exchange admixture of 20% in the well-known B3LYP functional by comparison to experimental data suggested to use a smaller exact exchange admixture of 15% (or less)13, 14 (see also ref.15 by Harvey for an up-to-date discussion). The performance of this B3LYP* functional for standard test sets is the same as for the original B3LYP functional.16 Consequently, the energies of the spin states of polynuclear clusters are also expected to vary strongly with the amount of exact exchange in the density functional. Because Heisenberg coupling constants are constructed as a measure for the energy difference between different spin states of a polynuclear transition metal cluster, they are also likely to be sensitive to exact exchange. In fact, dependencies of spin-state energy splittings17, 18 as well as of calculated coupling constants19-21 in dinuclear complexes on the density functional have been noticed in previous studies. But due to the lack of a systematic connection between different density functionals, no systematic investigation is possible. However, owing to the pronounced dependence of energy splittings on exact exchange, this Hartree–Fock-type contribution appears to be the only but promising contribution in the functionals to be investigated systematically. In this work we close the gap of a missing systematic study and conduct an investigation of the dependence of local spin expectation values and other quantities on the exact exchange admixture.
We aim at an overview of the energetic situation of all energetically close lying different spin states [not only the high-spin (hs) and Broken-Symmetry (BS) states] as predicted by DFT using contemporary density functionals. It is important to note that the term “different spin states” shall also refer not only to the total spin, but also to the spatial distribution of the spin excess in a Kohn–Sham determinant of given total spin. Because it is not known a priori which state is the ground state and which other states are close in energy and therefore may become important for the reactivity of the system under study (compare the two-state reactivity concept22), we must aim to converge all possible close lying spin states. For this it is necessary to screen a large part of the total energy hypersurface in the parameter space of molecular orbital (MO) coefficients. Therefore, we are adviced to investigate convergence of MO coefficients starting from low-quality guesses.
-
How does a modification of the exact exchange admixture affect the qualitative and quantitative properties of the converged SCF solutions?
-
How large is the influence of the exact exchange on the Heisenberg coupling constant calculated with three different mathematical expressions known from the literature? (The relation between these expressions shall also be explicated.)
-
How stable is the convergence of standard initial guesses for different total spin states with respect to to a varying exact exchange admixture?
As a test case, we have chosen a model for the intermediate Q in the MMOH reaction cycle, which has been proposed by Siegbahn23 (see also ref.24). This dinuclear complex is small enough to carry out a large number of comparative calculations. Furthermore, a large number of both experimental and theoretical results on MMO as well as on the structurally related Ribonucleotide Reductase (RNR)25, 26 has been published, which allows for comparisons to established density functionals, to other model systems,12, 27, 28 and to experiment. Quantum chemical studies on selected enzymes and catalytic synthetic systems in general and on MMO in particular have been recently reviewed by Noodleman et al.29 and by Baik et al.30 These reviews also include an overview on the reaction cycle of MMOH and the role of the intermediate Q, as well as extensive lists of references to the original articles.
This work is organized as follows: first, we briefly recall the theoretical background of spin contamination in Slater determinants, Clark and Davidson's local spin expectation values,31-34 and the calculation of Heisenberg coupling constants with Noodleman's BS approach35 to set the stage. In Sections 3 and 4, details on the computational methodology and the model cluster under study are presented. The following sections summarize total energies as well as total and local spin expectation values for different spin states. The results obtained employing standard density functionals are contained in Section 5, those obtained with B3LYP-based functionals with modified exact exchange admixture in Section 6. In Section 7, Heisenberg coupling constants calculated with expressions based on different assumptions on the interacting local spins and with different density functionals are investigated. A brief discussion of SCF convergence, some aspects of which are already adressed in Section 6, is summarized in Appendix A, which compares the convergence behavior of two standard ways of creating initial guess MOs for different spin states.
A Brief Review of the Theoretical Background
To make this account self-contained, to introduce the basic notation, and to review the basic facts of nonrelativistic open-shell Kohn–Sham (KS) DFT calculations, we decided to include a theoretical background section. This is also motivated by the fact that a discussion of spin–spin interactions often rests on a Hartree–Fock picture while the actual calculations are performed within DFT.
Spin Contamination in Unrestricted Kohn–Sham Determinants
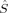 z〉 = MS value of MS = S can be adjusted in the initial guess determinant by choosing the appropriate number of α and β electrons, Nα and Nβ, because any Slater determinant |Ψ〉 is an eigenfunction of the
z〉 = MS value of MS = S can be adjusted in the initial guess determinant by choosing the appropriate number of α and β electrons, Nα and Nβ, because any Slater determinant |Ψ〉 is an eigenfunction of the
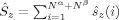 operator with the eigenvalue
operator with the eigenvalue
 ,
,
 (1)
(1)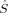 2〉 is in general larger than the desired value of MS(MS + 1). Second, the 〈
2〉 is in general larger than the desired value of MS(MS + 1). Second, the 〈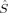 2〉 expectation value can be forced to be equal to MS(MS + 1) during the SCF iterations by applying the Lagrangian multiplier technique.36 This, however, leads in general to determinants with a higher energy than an unrestricted MO optimization without this constraint would yield.36 It can also lead to convergence problems in geometry optimizations. Slater determinants in Hartree–Fock theory should be eigenfunctions of the total spin operator, which commutes with the (spin-independent) nonrelativistic many-electron Hamiltonian. However, the Hamiltonian used in unrestricted KS-DFT, which depends on the spin density, describes a system of noninteracting fermions with same electronic density as the true electronic system. Because the KS surrogate system can exactly be described by a single Slater determinant (the potential energy terms are local), one may not request that Slater determinants used in KS-DFT should be eigenfunctions of
2〉 expectation value can be forced to be equal to MS(MS + 1) during the SCF iterations by applying the Lagrangian multiplier technique.36 This, however, leads in general to determinants with a higher energy than an unrestricted MO optimization without this constraint would yield.36 It can also lead to convergence problems in geometry optimizations. Slater determinants in Hartree–Fock theory should be eigenfunctions of the total spin operator, which commutes with the (spin-independent) nonrelativistic many-electron Hamiltonian. However, the Hamiltonian used in unrestricted KS-DFT, which depends on the spin density, describes a system of noninteracting fermions with same electronic density as the true electronic system. Because the KS surrogate system can exactly be described by a single Slater determinant (the potential energy terms are local), one may not request that Slater determinants used in KS-DFT should be eigenfunctions of 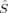 2. Pople, Gill, and Handy have advocated that unrestricted KS determinants should not be spin eigenfunctions, because they are no wave functions, that is, they are not the wave functions of fully interacting electronic systems.37 In a similar spirit, Perdew, Savin, and Burke argued that there is no obvious reason why the KS noninteracting wave function should always have the same symmetry as the true ground-state wave function.38 There appears to be no reason why the disadvantages of the spin-constrained MO optimization should be accepted in KS-DFT. Without this constraint, unrestricted KS determinants may (and must) exhibit spin contamination.
2. Pople, Gill, and Handy have advocated that unrestricted KS determinants should not be spin eigenfunctions, because they are no wave functions, that is, they are not the wave functions of fully interacting electronic systems.37 In a similar spirit, Perdew, Savin, and Burke argued that there is no obvious reason why the KS noninteracting wave function should always have the same symmetry as the true ground-state wave function.38 There appears to be no reason why the disadvantages of the spin-constrained MO optimization should be accepted in KS-DFT. Without this constraint, unrestricted KS determinants may (and must) exhibit spin contamination.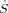 2〉 expectation value resulting from a quantum chemical calculation and the S(S + 1) eigenvalue, which is usually equal to MS(MS + 1) for the targeted S = MS determinants. The actual 〈
2〉 expectation value resulting from a quantum chemical calculation and the S(S + 1) eigenvalue, which is usually equal to MS(MS + 1) for the targeted S = MS determinants. The actual 〈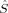 2〉 expectation value of a Slater determinant depends on the overlap integrals O = 〈ψ|ψ〉 of the spatial parts |ψ〉 and |ψ〉 of the α and β spin orbitals |ϕ〉 and |ϕ〉 (see, e.g., ref.39),
2〉 expectation value of a Slater determinant depends on the overlap integrals O = 〈ψ|ψ〉 of the spatial parts |ψ〉 and |ψ〉 of the α and β spin orbitals |ϕ〉 and |ϕ〉 (see, e.g., ref.39),
 (2)
(2)A high-spin determinant in the sense commonly used when talking about transition metal complexes and clusters, however, has as many excess α orbitals Nα – Nβ as there are valence electrons on the metal centers of the molecule it describes. Note that this is a qualitative picture taylored for the single-determinant wave functions that allows one to clearly identify those molecular orbitals with characteristic atomic d orbital contributions from the metal centers. Experience shows that such a determinant is in most cases not spin-contaminated. This can be explained by taking an idealized unrestricted determinant as an example, which may consist of “closed-shell” orbitals, indicating pairs of α and β spin orbitals with equal spatial parts, and where only the valence electrons of the metal centers are occupying orbitals whose spatial part does not (or only little) overlap with the spatial part of any orbital with the other spin. These orbitals are termed “magnetic orbitals” (for a rigorous definition of magnetic orbitals and how they can be obtained from the canonic orbitals by unitary transformations, see, e.g., refs.40-42). For the hs determinant, all valence electrons on the metal centers have the same spin (by convention: α). The sum
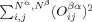 in eq. (2) then consists of Nβ contributions of one from the (normalized) spatially fully overlapping pairs of α and β orbitals, and Nα – Nβ contributions of zero from the magnetic α orbitals that do not overlap spatially with any β orbital. Note that the spatial parts of all β orbitals are equal to one of the other Nβα orbitals and the spatial parts of the α orbitals are by construction orthonormal among themselves. Thus, the sum is equal to Nβ, and the 〈
in eq. (2) then consists of Nβ contributions of one from the (normalized) spatially fully overlapping pairs of α and β orbitals, and Nα – Nβ contributions of zero from the magnetic α orbitals that do not overlap spatially with any β orbital. Note that the spatial parts of all β orbitals are equal to one of the other Nβα orbitals and the spatial parts of the α orbitals are by construction orthonormal among themselves. Thus, the sum is equal to Nβ, and the 〈 2〉 expectation value is equal to MS(MS + 1).
2〉 expectation value is equal to MS(MS + 1).
Such a qualitative picture might be close to reality, if either the unpaired electrons are occupying molecular orbitals with predominantly d atomic orbital character while the the core orbitals of the metals and the orbitals of the ligands keep their closed-shell character, or if the MOs can be transformed into a form close to this situation. This is confirmed by the fact that most hs (in the common, not in the strict sense) determinants are indeed not spin-contaminated.
Regarding the BS determinant, where the number of α and β electrons is the same and the total spin is equal to zero, while the local spins are not (the (nonzero) number of α electrons located at one metal atom is equal to the number of β electrons located at the other one), the sum in eq. (2) is lower than Nβ. In the extreme case the sum is equal to Nβ minus the number of unpaired α or β electrons. This is the number of “closed-shell” pairs of orbitals, whose overlap integrals each contribute a value of one to the sum, whereas the contributions from the overlap of the magnetic orbitals are much smaller than one. Therefore, spin contamination in unrestricted determinants is introduced by a lack of spatial overlap of pairs of α and β orbitals.
Heisenberg Model of Electron Spin Coupling
 (3)
(3) (4)
(4) (5)
(5) (6)
(6) (7)
(7) (8)
(8) 2〉 expectation value of the MS = 0 determinant. If all pairs of α and β orbitals overlap completely, the determinant is a closed-shell determinant, so BS〈
2〉 expectation value of the MS = 0 determinant. If all pairs of α and β orbitals overlap completely, the determinant is a closed-shell determinant, so BS〈 2〉 is equal to zero, and hs〈
2〉 is equal to zero, and hs〈 2〉 – BS〈
2〉 – BS〈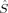 2〉 is equal to Shs(Shs + 1), the denominator in eq. (5). If there are magnetic orbitals that do not overlap at all (BS determinant), the sum in eq. (2) is a sum of Nβ – 2SA overlap integrals equal to one (the sum of unpaired electrons on A or B, and thus the number of magnetic orbitals of α or β spin, is equal to 2SA). Then the sum is equal to Nβ – 2SA and 〈
2〉 is equal to Shs(Shs + 1), the denominator in eq. (5). If there are magnetic orbitals that do not overlap at all (BS determinant), the sum in eq. (2) is a sum of Nβ – 2SA overlap integrals equal to one (the sum of unpaired electrons on A or B, and thus the number of magnetic orbitals of α or β spin, is equal to 2SA). Then the sum is equal to Nβ – 2SA and 〈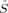 2〉 equals 2SA = Shs, and hs〈
2〉 equals 2SA = Shs, and hs〈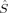 2〉 – BS〈
2〉 – BS〈 2〉 equals Shs(Shs + 1) – Shs = S, which is the denominator of eq. (4). Furthermore, Clark and Davidson's expression also reduces to Yamaguchi's expression in case that both the high-spin and the BS determinant are eigenfunctions of the local spin operators
2〉 equals Shs(Shs + 1) – Shs = S, which is the denominator of eq. (4). Furthermore, Clark and Davidson's expression also reduces to Yamaguchi's expression in case that both the high-spin and the BS determinant are eigenfunctions of the local spin operators 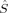 and
and 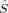 . Then, 2〈ŜA · ŜB〉 is equal to 〈
. Then, 2〈ŜA · ŜB〉 is equal to 〈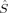 2〉 – SA(SA + 1) – SB(SB + 1) [see eq. (13)]. As mentioned above, the local spin quantum numbers SA and SB are equal for the hs and the BS determinant, so twice the difference between the 〈ŜA · ŜB〉 values of the hs and the BS determinant reduces to the difference of their total spin expectation values. This yields the denominator in Yamaguchi's expression, that is, both expressions are equal then.
2〉 – SA(SA + 1) – SB(SB + 1) [see eq. (13)]. As mentioned above, the local spin quantum numbers SA and SB are equal for the hs and the BS determinant, so twice the difference between the 〈ŜA · ŜB〉 values of the hs and the BS determinant reduces to the difference of their total spin expectation values. This yields the denominator in Yamaguchi's expression, that is, both expressions are equal then.Local Spin
 (9)
(9) (10)
(10) (11)
(11)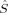 zA and NA to the z-component of the total spin operator,
zA and NA to the z-component of the total spin operator, 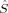 z, and to the total electron number N, can also be formulated in terms of these local projection operators,51 as has been first proposed for population analyses by Clark and Davidson.52 The expectation values of the local z-component
z, and to the total electron number N, can also be formulated in terms of these local projection operators,51 as has been first proposed for population analyses by Clark and Davidson.52 The expectation values of the local z-component 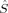 zA of the total spin, and the local population of α and β electrons, N and N, are related to the local 〈
zA of the total spin, and the local population of α and β electrons, N and N, are related to the local 〈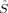 zA〉 value, which is half the spin density on center A,
zA〉 value, which is half the spin density on center A,
 (12)
(12)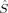 zA values close to those obtained with the formally appealing Atoms-in-Molecules projectors55 for small molecules.51 It is therefore employed in this work for the calculation of the local spin expectation values 〈
zA values close to those obtained with the formally appealing Atoms-in-Molecules projectors55 for small molecules.51 It is therefore employed in this work for the calculation of the local spin expectation values 〈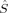 zA〉 and 〈ŜA · ŜB〉. A derivation of expressions for these expectation values is given in refs.34 and51. Whereas the 〈ŜA · ŜB〉 expectation value is useful for the calculation of coupling constants, the 〈
zA〉 and 〈ŜA · ŜB〉. A derivation of expressions for these expectation values is given in refs.34 and51. Whereas the 〈ŜA · ŜB〉 expectation value is useful for the calculation of coupling constants, the 〈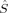 zA〉 expectation value can be used for assigning local spin states to metal atoms in transition metal complex.
zA〉 expectation value can be used for assigning local spin states to metal atoms in transition metal complex.Calculation of Ideal Local Spin Values
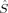 2 and of the local spin operators
2 and of the local spin operators 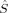 and
and 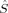 . Then, its ideal local spin eigenvalue 〈ŜA · ŜB〉 can be calculated from
. Then, its ideal local spin eigenvalue 〈ŜA · ŜB〉 can be calculated from
 (13)
(13) (14)
(14) (15)
(15)Computational Methods
All-electron calculations were performed with the quantum chemical program package TURBOMOLE56, 57 employing either the BP86 functional58, 59 in combination with the resolution-of-the-identity (RI) density fitting technique,60, 61 (ftp://ftp.chemie.uni-karlsruhe.de/pub/jbasen) the B3LYP functional,62, 63 or a modified B3LYP functional using a modified implementation of TURBOMOLE's DSCF module. In the modified B3LYP functional, the exact exchange admixture is varied from 0 to 24%. It is equal to 20% in the original B3LYP functional, and equal to 15% in the case of the B3LYP* functional.13, 14, 16 We used the TZVP basis by Schäfer et al. (ftp://ftp.chemie.uni-karlsruhe.de/pub/basen) throughout, which features a valence triple-zeta basis set with polarization functions on all atoms. Test calculations with Ahlrichs' TZVPP basis set, which comprises two Dunning-type polarization functions on all atoms, showed only minor changes with respect to the TZVP results. For the calculation of local spin expectation values, we applied our implementation described in ref.51 in TURBOMOLE. For the local decomposition, modified Löwdin projectors,31 denoted by Löwdin*, were used. Extended Hückel MOs were obtained from TURBOMOLE's DEFINE tool. Molecular structures were visualized with the program MOLDEN.64
The MMOH Model as an Example
The model for the active center of the intermediate Q in the MMOH reaction cycle, which is investigated in this work follows the structure proposal of Siegbahn23 (see Fig. 1). It consists of a diiron center with formal oxidation numbers of IV, bridged by two oxygen atoms and by two acetate ions, which model a glutamic acid and an aspartate residue. Each iron center is furthermore surrounded by another, unidentate acetate ion and by an imidazole ligand, replacing a glutamic acid and a histidine residue, respectively. Consequently, each iron center possesses six ligands.

Structure of the MMO model complex, geometry optimized with B3LYP/TZVP (2) for the hs (S = 4) state. Selected bond distances are given in Ångstroms. The values in parentheses refer to the BP86/TZVP optimized structure (1).
Experimental Results for MMOH
As far as they are relevant for our calculations, the experimental results known for the intermediate Q of MMOH shall be summarized here. These data only serve as an orientation, because differences between calculated and measured values may originate for various reasons.
It is known from Mössbauer spectroscopy that there are two antiferromagnetically coupled iron(IV) atoms in Q with a coupling constant of JFe1Fe2 lower than −30cm−1.65, 66 Furthermore, according to EXAFS spectra, the iron–oxygen bridges are asymmetric with Fe–O distances of 1.77 and 2.06 Å, and the Fe–Fe distance is between 2.46 and 2.52 Å.67, 68 XAS data propose a coordination number of five or lower for the iron atoms.67 For more details on how the geometry of Siegbahn's model compares to experiment see ref.29.
Possible Spin States Obtained by Chemical Intuition
The two iron centers of the MMO model complex have the formal oxidation number IV, which corresponds to a d4 configuration. The total spin state, which is usually referred to as hs is therefore the one with four unpaired electrons on each of the two iron atoms, which all have the same spin, yielding a total S quantum number of 4. Because the total number of electrons in the model complex is even, the lowest possible spin state is characterized by S = 0. This total spin can result from several local spin distributions. The two extrema are an equal distribution of the α and the β spin density over the molecule (closed-shell), and two locally hs iron centers that are coupled antiferromagnetically (MSFe = ±2). The latter case is represented by one KS determinant here, which corresponds to Noodleman's BS solution.35
The two determinants that represent local hs states, that is, the BS and the total hs determinant, correspond to states that can be described by a Heisenberg model with two interacting S = 2 spins. If the energy of other spin states, however, is close enough to the ground state, they should not be neglected. Therefore, we also consider intermediate spin states.
Construction of Slater Determinants and Notation
To achieve the goal to present a complete picture of the energetics of relevant spin states as predicted by DFT, the full range of low-, intermediate-, and high-spin states has to be converged. We are aware that usually only the BS and hs states are converged by setting up a quite sophisticated MO guess, which then may converge more quickly to the states with desired total and local spin properties. However, there is no formal reason why fast convergence may always be expected. Therefore, we investigate convergence from standard initial guesses. It should be emphasized, however, that all states obtained after the SCF optimization are, of course, stationary points in the parameter space of the MO coefficients used for the calculation of the total electronic energy. A BS-like guess may only converge faster to some of these solutions.
Two different standard initial guesses for the MOs have been employed. MOs obtained from an Extended Hückel calculation (E) and the converged KS orbitals of the hs (S = 4) state (H) using the same functional and structure as for the actual calculation with lower spin. Both initial guesses have been used for calculating MOs with the BP86, the standard B3LYP, and modified B3LYP functionals with a varying fraction of exact exchange, for a BP86 optimized (1) and a B3LYP optimized (2) hs structure. The resulting determinants have been labeled by a special notation that is explained in Table 1.
| Symbol | Description |
|---|---|
| 1 | Optimized structure with BP86/RI/TZVP |
| 2 | Optimized structure with B3LYP/TZVP |
| R | restricted-(R)KS-DFT(closed-shell) calculation |
| E | Extended Hückel initial guess MOs |
| H | High spin state initial guess molecular orbitals (S = 4), obtained with the same functional and structure as used in the actual calculation |
| * | Converged MOs of 1(S1,H,B3LYP) as initial guess |
| B | Broken-Symmetry (BS) (S = 0) initial guess MOs obtained with B3[0.04] |
| Si | Spin state with total spin quantum number S=i |
| B3[c3] | Modified B3LYP density functional with exact exchange admixture parameter c3. |
| BP | BP86 density functional |
- The symbol 2(S0, RE, B3[0.15]) for example refers to a restricted KS-DFT calculation was performed using the B3LYP/TZVP optimized structure 2 for a closed-shell singlet based on Extended Hückel start MOs and using a modified B3LYP density functional with 15% exact exchange admixture. If not denoted explicitely by the symbol R, all calculations have been performed within unrestricted DFT.
Optimized Cluster Structures
To evaluate the influence of the density functional on the geometry, and the influence of this geometry difference onto the spin state energies, the molecular structures optimized with two functionals are compared. These are the pure density functional BP86 and the hybrid density functional B3LYP, which contains 20% of exact exchange. The triple-zeta basis set with polarization functions TZVP was employed. Because the choice of the density functional can influence the local spin properties of the KS determinant to which a given initial guess converges in the SCF algorithm, the hs state (S = 4), which features only one chemically reasonable local spin distribution (M = M = S/2), has been chosen for the optimization. The conclusions on the influence of the exact exchange in the density functional and the SCF convergence of standard initial guesses can be expected not to be affected by using structures obtained by optimization of other spin states. The optimization has been carried out in C1 symmetry. The optimized molecular structures of the MMO model obtained with B3LYP and BP86 (values in parentheses) are compared in Figure 1.
Whereas the Fe–Fe distance is the same in both structures, the symmetry of the oxygen bridge depends on the density functional. The Fe–O bond lengths to Fe1 and Fe2 differ by 0.197 and 0.166 Å for the bridging oxygen atoms O1 and O2, respectively, in the B3LYP optimized structure. The result is a rhomboid-like geometry for the quadrangle consisting of Fe1, O1, Fe2, and O2. In contrast to this, in the BP86 optimized structure all Fe–O distances are equal and about in the middle between the two different values for the B3LYP structure. They range from 1.802 to 1.872 Å, differing thus by at most 0.025 Å. Because the geometry optimization has been performed in C1 symmetry, that is, without symmetry, the structures are not perfectly symmetric. However, this slight distortion of the perfect symmetry is in the spirit of this work as it allows us to search for two BS solutions of slightly different energy.
Compared to experiment, the B3LYP optimized structure reproduces the asymmetric oxygen bridge quite well, whereas the BP86 structure does not. For both functionals, the iron–iron distance is about 0.1 to 0.2 Å longer than experiments suggest. One might expect this to change when using antiferromagnetically coupled BS MOs for the geometry optimization, because Mössbauer data indicate that this is the ground state. However, a structure optimization with the hs geometries as starting point yielded a geometry even further away from experiment with iron–iron distances of 2.610 (2.590) Å and oxygen bridges which are symmetric with respect to the bridge, but asymmetric with respect to on which side the bridge is. On the imidazole side, the Fe–O distances turned out to be 1.806 (1.800) and 1.800 (1.802) Å, whereas on the acetate side, they are equal to 1.773 (1.763) and 1.777 (1.763) Å. The values for B3LYP and BP86 (in parentheses) do not differ much. Siegbahn obtained structural parameters much closer to experiment23 for this model with B3LYP. This might be either due to different stationary points on the potential energy surface reached in this and in Siegbahn's work, or to the smaller double-zeta basis set and the ECP Siegbahn employed. We have chosen the hs geometry obtained with B3LYP for all further investigations of the spin state energies and spin properties, because it is closest to the experimental data. There is no reason to expect large deviations for the dependency of the SCF convergence behavior and the spin state energetics on the functional when changing the geometry for a few tenth of an Ångstrøm.
Energies and Spin Expectation Values of Different Spin States with Standard Functionals
Total Energies
The total energies obtained with KS-DFT for different total spin states using the standard density functionals BP86 and B3LYP are summarized in Figure 2. The energy of the closed-shell singlet determinant obtained with an Extended Hückel guess and the corresponding density functional and structure is taken as the reference energy.

Relative total energies ΔE of the MMO model complex in kJ/mol for different structures and functionals, obtained with two different sets of initial guess MOs, denoted by E and H (see Table 1 for an explanation). The energy of the closed-shell singlet determinant obtained with a Extended Hückel guess (S0, RE) and the corresponding density functional and structure is taken as reference. In cases where both initial guesses have converged to a solution where the local spins on the iron atoms are the same or just interchanged or flipped, and the energy difference is less than 4 kJ/mol, only one of them (E) is given. The determinants 1(S1,H,BP) and 1(S2,H,B3) have not been included in Figure 2, which differ from the corresponding “E” determinants only by having the local spins on the iron atoms interchanged. This interchange results in energy differences of 3.7 and 3.5 kJ/mol, respectively, which is due to the fact that the geometry optimization has been performed in C1 symmetry, and that the two iron atoms are therefore not strictly symmetry equivalent.
Figure 2 shows that the absolute values of the total energies depend strongly on the functional. The BP86 energies for a given structure are spread over a range of at most 155 kJ/mol, whereas for the B3LYP energies, the range is more than twice as large (up to 341 kJ/mol). The order of the spin states on the energy scale is the same for the following determinants regardless of functional and geometry: the BS determinant (with S = 0) has always the lowest energy and the restricted closed-shell determinant the highest one. In between, there are always a S = 1, a S = 3 and a S = 2 determinant with the same local spin properties for both functionals and both structures, and with the energy increasing in this order. The hs determinant, which has the highest energy of all unrestricted determinants when calculated with the pure functional BP86, is favored so much in the hybrid functional B3LYP calculation that it has the second-lowest energy after the BS determinant. For a more detailed analysis, knowledge on the local spins is necessary, so the discussion will be continued in the next section.
Local Spins
A comparison of total energies is only sensible if the corresponding determinants optimized with different density functionals describe the same electronic structure, that is, possess similar local spins. The relevant total and local spin expectation values of all determinants obtained with the different initial guesses, geometries and functionals are summarized in Table 2. These are the total spin expectation value 〈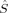 2〉, which is equal to S(S + 1) for total spin eigenfunctions, and the portion of the z-component of the total spin that can be attributed to the iron centers and to the bridging oxygen atoms,
2〉, which is equal to S(S + 1) for total spin eigenfunctions, and the portion of the z-component of the total spin that can be attributed to the iron centers and to the bridging oxygen atoms, 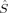 zFe1,
zFe1, 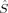 zFe2,
zFe2, 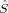 zO1, and
zO1, and  zO2. The 〈ŜFe1 · ŜFe2〉 expectation value is also listed, which will be needed below for the calculation of Heisenberg coupling constants according to the equation by Clark and Davidson. The values for the closed-shell determinants have not been included, since their total and local spin expectation values (except 〈ŜFe1 · ŜFe2〉) are all equal to zero. The Löwdin* local projection scheme was used throughout for formal reasons (see earlier). Although usually employed in the literature, the (not rigorously defined) corresponding Mulliken local 〈
zO2. The 〈ŜFe1 · ŜFe2〉 expectation value is also listed, which will be needed below for the calculation of Heisenberg coupling constants according to the equation by Clark and Davidson. The values for the closed-shell determinants have not been included, since their total and local spin expectation values (except 〈ŜFe1 · ŜFe2〉) are all equal to zero. The Löwdin* local projection scheme was used throughout for formal reasons (see earlier). Although usually employed in the literature, the (not rigorously defined) corresponding Mulliken local 〈 zA〉 values are not reported because they are very close to the Löwdin* ones. This is different from population analysis, where the Löwdin and the Mulliken partitioning scheme may yield quite different results.69, 70 Obviously, the dependence of the absolute α and β populations on the projection operator cancel for the example under study here when differences between those absolute populations are considered. As far as the local decomposition of the
zA〉 values are not reported because they are very close to the Löwdin* ones. This is different from population analysis, where the Löwdin and the Mulliken partitioning scheme may yield quite different results.69, 70 Obviously, the dependence of the absolute α and β populations on the projection operator cancel for the example under study here when differences between those absolute populations are considered. As far as the local decomposition of the  2 operator is concerned, on the other hand, no standard local projection operator has been established so far. Because their dependence on the local projection scheme has already been investigated,31-33,51 we restrict this work to the formally consistent Löwdin* projectors for the calculation of 〈ŜFe1 · ŜFe2〉 values.
2 operator is concerned, on the other hand, no standard local projection operator has been established so far. Because their dependence on the local projection scheme has already been investigated,31-33,51 we restrict this work to the formally consistent Löwdin* projectors for the calculation of 〈ŜFe1 · ŜFe2〉 values.
| Spin state | Functional | 〈 2〉 2〉 |
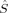 zFe1 zFe1 |
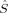 zFe2 zFe2 |
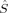 zO1 zO1 |
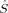 zO2 zO2 |
〈 Fe1· Fe1· Fe2〉 Fe2〉 |
|---|---|---|---|---|---|---|---|
| S4 E | 1 BP86 | 20.06 | 1.55 | 1.56 | 0.23 | 0.17 | 2.30 |
| 1 B3LYP | 20.13 | 1.67 | 1.68 | 0.18 | 0.11 | 2.76 | |
| 2 BP86 | 20.06 | 1.52 | 1.55 | 0.26 | 0.18 | 2.32 | |
| 2 B3LYP | 20.13 | 1.66 | 1.70 | 0.20 | 0.12 | 2.70 | |
| S3 E | 1 BP86 | 12.10 | 1.35 | 1.08 | 0.19 | 0.18 | 1.31 |
| 1 B3LYP | 12.19 | 1.04 | 1.62 | 0.14 | 0.11 | 1.62 | |
| 2 BP86 | 12.07 | 1.47 | 0.89 | 0.18 | 0.20 | 1.26 | |
| 2 B3LYP | 12.13 | 1.63 | 0.93 | 0.14 | 0.17 | 1.40 | |
| S3 H | 1 BP86 | 12.10 | 1.34 | 1.09 | 0.19 | 0.18 | 1.31 |
| 1 B3LYP | 12.19 | 1.04 | 1.61 | 0.15 | 0.11 | 1.62 | |
| 2 BP86 | 12.07 | 1.47 | 0.89 | 0.17 | 0.20 | 1.26 | |
| 2 B3LYP | 12.13 | 1.63 | 0.93 | 0.14 | 0.16 | 1.40 | |
| S2 E | 1 BP86 | 6.12 | 0.81 | 0.81 | 0.17 | 0.14 | 0.44 |
| 1 B3LYP | 6.55 | 0.67 | 1.02 | 0.24 | 0.10 | 0.62 | |
| 2 BP86 | 6.18 | 0.80 | 0.82 | 0.17 | 0.16 | 0.54 | |
| 2 B3LYP | 6.37 | 0.80 | 0.87 | 0.20 | 0.17 | 0.58 | |
| S2 H | 1 BP86 | 6.12 | 0.81 | 0.81 | 0.17 | 0.14 | 0.44 |
| 1 B3LYP | 6.62 | 1.09 | 0.62 | 0.25 | 0.06 | 0.61 | |
| 2 BP86 | 6.18 | 0.80 | 0.81 | 0.17 | 0.15 | 0.54 | |
| 2 B3LYP | 7.03 | 1.64 | −0.10 | 0.13 | 0.13 | −0.28 | |
| S1 E | 1 BP86 | 3.67 | −0.68 | 1.41 | 0.16 | −0.03 | −1.12 |
| 1 B3LYP | 3.15 | 0.56 | 0.46 | 0.34 | −0.33 | 0.20 | |
| 2 BP86 | 3.97 | −0.70 | 1.44 | −0.10 | −0.15 | −1.09 | |
| 2 B3LYP | 3.48 | −0.31 | 0.90 | 0.30 | 0.15 | −0.4 | |
| S1 H | 1 BP86 | 3.68 | 1.42 | −0.68 | 0.17 | −0.04 | −1.13 |
| 1 B3LYP | 4.14 | 1.65 | −0.97 | 0.15 | −0.05 | −1.65 | |
| 2 BP86 | 3.75 | 1.44 | −0.70 | 0.20 | −0.10 | −1.09 | |
| 2 B3LYP | 3.23 | 0.77 | −0.28 | 0.26 | 0.26 | −0.34 | |
| *2 B3LYP | 4.09 | 1.64 | −0.89 | 0.17 | −0.12 | −1.51 | |
| S0 E | 1 BP86 | 3.33 | −1.41 | 1.42 | 0.01 | −0.01 | −2.19 |
| 1 B3LYP | 2.22 | 0.39 | 0.35 | −0.35 | −0.32 | 0.08 | |
| 2 BP86 | 3.38 | 1.39 | −1.41 | 0.09 | −0.09 | −2.07 | |
| 2 B3LYP | 2.21 | −0.37 | 0.33 | 0.33 | −0.29 | −0.24 | |
| S0 H | 1 BP86 | 3.33 | −1.42 | 1.42 | −0.01 | 0.01 | −2.19 |
| 1 B3LYP | 2.19 | −0.38 | −0.35 | 0.35 | 0.32 | 0.08 | |
| 2 BP86 | 3.38 | −1.39 | 1.41 | −0.08 | 0.09 | −2.07 | |
| 2 B3LYP | 2.30 | −0.34 | −0.29 | 0.32 | 0.25 | −0.02 |
Ideal Local Spin Values
To be able to judge to which degree the determinants are close to eigenfunctions of the total and local spin operators, the ideal values for the corresponding expectation values are given in Table 3 for a selection of ideal spin states that are closest to the values in Table 2. The ideal 〈ŜFe1·ŜFe2〉 expectation values are calculated according to eq. (13). The total 〈 z〉 expectation values are not included in Tables 2 and 3, because all Slater determinants are eigenvalues of
z〉 expectation values are not included in Tables 2 and 3, because all Slater determinants are eigenvalues of  z by construction. The states which are obtained by interchanging the ideal 〈
z by construction. The states which are obtained by interchanging the ideal 〈 zFe〉 values are not included either, because the labeling of the iron atoms is not essential.
zFe〉 values are not included either, because the labeling of the iron atoms is not essential.
| Spin state | 〈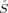 2〉 2〉 |
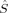 zFe1 zFe1 |
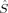 zFe2 zFe2 |
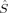 zO1 zO1 |
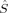 zO2 zO2 |
〈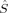 zFe1 zFe1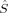 zFe2〉 zFe2〉 |
|
|---|---|---|---|---|---|---|---|
| S4 | 20.00 | 2.00 | 2.00 | 0.00 | 0.00 | 4.00 | ↑↑↑↑ - ↑↑↑↑ |
| S3 | 12.00 | 2.00 | 1.00 | 0.00 | 0.00 | 2.00 | ↑↑↑↑ - ↑↑ |
| S2 | 6.00 | 2.00 | 0.00 | 0.00 | 0.00 | 0.00 | ↑↑↑↑ - 0 |
| 6.00 | 1.00 | 1.00 | 0.00 | 0.00 | 1.00 | ↑↑ - ↑↑ | |
| S1 | 2.00 | 2.00 | −1.00 | 0.00 | 0.00 | −3.00 | ↑↑↑↑ - ↓↓ |
| 2.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | ↑↑ - 0 | |
| S0 | 0.00 | 2.00 | −2.00 | 0.00 | 0.00 | −6.00 | ↑↑↑↑ - ↓↓↓↓ |
| 0.00 | 1.00 | −1.00 | 0.00 | 0.00 | −2.00 | ↑↑ - ↓↓ |
- In the last column, the local spins on the iron atoms are depicted. Excess α and β electrons are marked by arrows pointing up and down, respectively.
Drawing conclusions from local spin expectation values on local SA quantum numbers in KS determinants within the collinear KS-DFT applied here is not straightforward. The 〈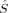 A2〉 expectation values yield nonzero local spin quantum numbers for closed-shell molecules with zero spin density at every point of space,51 and are therefore not a reliable measure for the local spin quantum numbers SA on the magnetic centers for any molecule that contains a “closed-shell” contribution to chemical bonds of the magnetic centers. Note that this case cannot be excluded for the transition metal complex under investigation here. The local 〈
A2〉 expectation values yield nonzero local spin quantum numbers for closed-shell molecules with zero spin density at every point of space,51 and are therefore not a reliable measure for the local spin quantum numbers SA on the magnetic centers for any molecule that contains a “closed-shell” contribution to chemical bonds of the magnetic centers. Note that this case cannot be excluded for the transition metal complex under investigation here. The local 〈 zA〉 expectation values describe closed-shell molecules in accordance with chemical intuition, but the local M quantum numbers they yield are not necessarily equal to the local SA quantum numbers. However, in accordance with total spin quantum numbers of Slater determinants, where we use MS as a means to produce a well-defined total spin quantum number for S, we will interpret the 〈
zA〉 expectation values describe closed-shell molecules in accordance with chemical intuition, but the local M quantum numbers they yield are not necessarily equal to the local SA quantum numbers. However, in accordance with total spin quantum numbers of Slater determinants, where we use MS as a means to produce a well-defined total spin quantum number for S, we will interpret the 〈 zA〉 values as local SA quantum numbers.
zA〉 values as local SA quantum numbers.
Assignment of Ideal Local Spin Values
In Table 2, the local spin excess has been assumed to be distributed onto the two iron atoms only. Because there are four unpaired electrons attributed to each iron center, the possible local spin quantum numbers SFe are 0, ±1 and ±2. The local 〈 zA〉 values show that the spin excess is not exclusively located on the iron centers, but also on the bridging oxygen atoms. By assuming that this delocalized spin would be localized on the iron centers in the ideal states, most of the KS determinants listed in Table 2 can be assigned to one of the ideal determinants in Table 3.
zA〉 values show that the spin excess is not exclusively located on the iron centers, but also on the bridging oxygen atoms. By assuming that this delocalized spin would be localized on the iron centers in the ideal states, most of the KS determinants listed in Table 2 can be assigned to one of the ideal determinants in Table 3.
The hs (S = 4) determinants all correspond to two ferromagnetically coupled locally hs Fe centers, with a spin delocalization onto the bridging oxygen atoms that reduces the 〈 zFe〉 to values between 1.52 and 1.70 instead of the ideal value of 2.
zFe〉 to values between 1.52 and 1.70 instead of the ideal value of 2.
For the septet (S = 3) determinants, both initial guesses have always converged to the same local spin state with 〈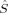 zFe1〉 = 1 and 〈
zFe1〉 = 1 and 〈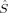 zFe2〉 = 2 (or vice versa). Its energy is always between the lower one of the triplet states and the lowest quintet state.
zFe2〉 = 2 (or vice versa). Its energy is always between the lower one of the triplet states and the lowest quintet state.
The quintet (S = 2) determinants all have 〈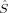 zFe1〉 = 1 and 〈
zFe1〉 = 1 and 〈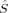 zFe2〉 = 1, except 2(S2,H,B3), which corresponds to a state where all spin excess is located on one of the iron atoms with 〈
zFe2〉 = 1, except 2(S2,H,B3), which corresponds to a state where all spin excess is located on one of the iron atoms with 〈 zFe1〉 = 2 and 〈
zFe1〉 = 2 and 〈 zFe2〉 = 0. The latter is 23.1 kJ/mol lower in energy than the former according to the B3LYP calculations on structure 2, but more difficult to converge. The amount of spin delocalization onto the bridging oxygen atoms is comparable for all the septet and quintet determinants and ranges from 〈
zFe2〉 = 0. The latter is 23.1 kJ/mol lower in energy than the former according to the B3LYP calculations on structure 2, but more difficult to converge. The amount of spin delocalization onto the bridging oxygen atoms is comparable for all the septet and quintet determinants and ranges from 〈 zO〉 = 0.06 to 〈
zO〉 = 0.06 to 〈 zO〉 = 0.26.
zO〉 = 0.26.
In the case of S=1 (triplet), most determinants can be described by 〈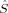 zFe1〉 = 2 and 〈
zFe1〉 = 2 and 〈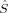 zFe2〉 = −1 (or vice versa). The local spin expectation values on the bridging oxygen atoms can be positive or negative and range from 〈
zFe2〉 = −1 (or vice versa). The local spin expectation values on the bridging oxygen atoms can be positive or negative and range from 〈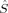 zO〉 = −0.15 to 〈
zO〉 = −0.15 to 〈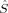 zO〉 = 0.20. These determinants are always comparatively low in energy. The BP86 determinant 2(S1,H,BP) does not converge to the same state as the S = 1 high-spin initial guess for the other three combinations of geometry and functional, but to a higher energy state with the local spins on iron interchanged. The reasons for this might be that the spin inversion on the iron atoms is paralleled by a qualitative change of the spin delocalization onto the oxygen bridge, which is possible because both iron atoms are not exactly symmetry equivalent. In the higher energy solution, which was obtained from the hs MO guess, one oxygen atom has a positive and the other one a negative spin density, whereas in the lower energy determinant obtained from the Extended Hückel guess, both have negative spin densities.
zO〉 = 0.20. These determinants are always comparatively low in energy. The BP86 determinant 2(S1,H,BP) does not converge to the same state as the S = 1 high-spin initial guess for the other three combinations of geometry and functional, but to a higher energy state with the local spins on iron interchanged. The reasons for this might be that the spin inversion on the iron atoms is paralleled by a qualitative change of the spin delocalization onto the oxygen bridge, which is possible because both iron atoms are not exactly symmetry equivalent. In the higher energy solution, which was obtained from the hs MO guess, one oxygen atom has a positive and the other one a negative spin density, whereas in the lower energy determinant obtained from the Extended Hückel guess, both have negative spin densities.
The four S = 0 determinants obtained with BP86 all correspond to a BS solution with absolute values of the local spins on the iron atoms between 1.39 and 1.42. This corresponds most likely to local spins of 〈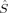 zFe1〉 = 2 and 〈
zFe1〉 = 2 and 〈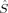 zFe2〉 = −2, because the local 〈
zFe2〉 = −2, because the local 〈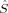 zFe〉 values of the hs determinant are also diminished by spin delocalization. However, the local spins do not allow for an unambiguous differentiation from 〈
zFe〉 values of the hs determinant are also diminished by spin delocalization. However, the local spins do not allow for an unambiguous differentiation from 〈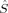 zFe1〉 = 1 and 〈
zFe1〉 = 1 and 〈 zFe2〉 = −1 (or vice versa). The spin delocalization onto oxygen is small (between 〈
zFe2〉 = −1 (or vice versa). The spin delocalization onto oxygen is small (between 〈 zO〉 = −0.09 and 〈
zO〉 = −0.09 and 〈 zO〉 = 0.09). The BS determinant has the lowest energy in all cases.
zO〉 = 0.09). The BS determinant has the lowest energy in all cases.
It is important to note that three of the five B3LYP/S = 1 determinants, 1(S1,E,B3), 2(S1,E,B3), and 2(S1,H,B3), and all four B3LYP/S = 0 determinants, 1(S0,E,B3), 2(S0,E,B3), 1(S0,H,B3), and 2(S0,H,B3), cannot be attributed to any of the ideal local spin states. All of them exhibit a spin delocalization onto the oxygen bridges, which is around |〈 zO〉| = 0.30 and thus rather large compared to the small total spin. This spin distribution is not consistent with any chemically sensible qualitative picture of the electron configuration for a transition metal complex. The high energy of these seven determinants (see Fig. 2) suggests that they correspond to local minima to which the SCF algorithm has converged, and not to the chemically desired states. By taking the converged MOs of a reasonable triplet determinant like 1(S1,H,B3) as an initial guess for a B3YLP calculation on structure 2, one obtains the low-energy determinant 2(S1,*,B3) with the same local spin properties as the initial guess instead of the chemically unreasonable determinants 2(S1,E,B3) and 2(S1,H,B3).
zO〉| = 0.30 and thus rather large compared to the small total spin. This spin distribution is not consistent with any chemically sensible qualitative picture of the electron configuration for a transition metal complex. The high energy of these seven determinants (see Fig. 2) suggests that they correspond to local minima to which the SCF algorithm has converged, and not to the chemically desired states. By taking the converged MOs of a reasonable triplet determinant like 1(S1,H,B3) as an initial guess for a B3YLP calculation on structure 2, one obtains the low-energy determinant 2(S1,*,B3) with the same local spin properties as the initial guess instead of the chemically unreasonable determinants 2(S1,E,B3) and 2(S1,H,B3).
As far as the closed-shell determinants are concerned, the fact that they are always the very highest in energy underlines that restricted KS-DFT is not a good description for this system. Both initial guesses converge to the same solution, except the B3LYP determinants obtained for structure 2, which differ by 10.9 kJ/mol in energy. Because second derivatives of the KS energy expression with respect to the MO coefficients are not readily available, we cannot rule out whether this is because 2(S0,RE,B3) is a saddlepoint or a higher energy local minimum.
These data demonstrate that especially for the lower spin state, SCF convergence is not easily achieved by standard means. This holds in particular for the hybrid functional B3LYP, whereas for the pure functional BP86 convergence is more stable. Furthermore, for the B3LYP optimized structure 2, which has antisymmetric oxygen bridges, the probability that Extended Hückel and the hs guesses converge to different solutions is higher than for the BP86 optimized structure 1 with symmetric oxygen bridges.
Total Spin
The spin contamination is, in general, low for the higher spin states (S = 2 to S = 4), but increases the more the local 〈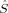 zA〉 values on the two iron atoms differ. Such differences occur more frequently for the lower-spin states (S = 1 and S = 0). The 2(S2,H,B3) determinant with ideal local iron spins of 2 and 0, for example, has a 〈
zA〉 values on the two iron atoms differ. Such differences occur more frequently for the lower-spin states (S = 1 and S = 0). The 2(S2,H,B3) determinant with ideal local iron spins of 2 and 0, for example, has a 〈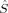 2〉 expectation value of 7.03, whereas the same determinant obtained from an Extended Hückel guess, 2(S2,E,B3), with ideal spins of 1 on both iron atoms, has only 6.37, which is fairly close to the ideal value of 6. The BS determinants (with S = 0), with 〈
2〉 expectation value of 7.03, whereas the same determinant obtained from an Extended Hückel guess, 2(S2,E,B3), with ideal spins of 1 on both iron atoms, has only 6.37, which is fairly close to the ideal value of 6. The BS determinants (with S = 0), with 〈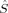 2〉 between 3.33 and 3.38, all deviate strongly from the ideal total spin expectation value of zero. For determinants with comparable total and local spins, the hybrid functional B3LYP always yields a slightly higher 〈
2〉 between 3.33 and 3.38, all deviate strongly from the ideal total spin expectation value of zero. For determinants with comparable total and local spins, the hybrid functional B3LYP always yields a slightly higher 〈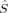 2〉 expectation value.
2〉 expectation value.
Dependence of Energies and Spins on the Exact Exchange Admixture
 (16)
(16)Total Energies
The dependence of the total energies of the dinuclear MMO model complex on the exact exchange admixture in the hs B3LYP optimized geometry (structure 2) is depicted in Figure 3.

Figure 3 is divided into two plots. The upper one contains results for all determinants whose energy depends continuously on c3 (including the higher spin states with S = 4, S = 3, and S = 2). The lower one contains those determinants for which the energy plot is discontinuous (with the lower spins S = 1 and S = 0). The discontinuities indicate qualitative changes in the unrestricted determinants (the reference closed-shell state possesses, of course, a perfectly continuous behavior with varying c3). If the initial guess was modified appropriately and/or the state to which the SCF algorithm converges could be controlled during the MO optimization, it might be possible to obtain several continuous lines corresponding to all these different states as indicated by dotted lines in Figure 3. For the S = 0 state, this continuation is carried out explicitely by taking the molecular orbitals from the determinant 2(S0,H,B3[0.04]) as an initial guess for electronic structure calculations with S = 0 and with c3 parameters ranging from 0.08 to 0.24. This determinant was chosen because it is closest to the lower energy side of the discontinuity of the 2(S0,H) plot.
 (17)
(17)In addition to the absolute energies, the relative values for different spin states also change when c3 is varied. The hs (S = 4) determinant possesses the highest energy of the four determinants plotted in the upper part of Figure 3 when the exact exchange admixture is equal to zero, but has the lowest energy of the four when the exact exchange admixture is higher than 12%. The BS determinant 2(S0 HB/EB) in the lower part, which depends continuously on c3, has always the lowest energy of all spin states under investigation. Furthermore, Figure 3 shows that the energies cover a broader range of values when the c3 parameter is increased. These qualitative findings are in agreement with the corresponding energy scheme for the pure BP86 functional and the original B3LYP functional in Figure 2.
Furthermore, it can be concluded that to which state a determinant converges strongly depends on the exact exchange admixture in the density functional when the total spin state is low (S = 0 or S = 1). These determinants obtained with either Extended Hückel or hs guess MOs suddenly converge to higher-energy states when c3 is higher than 0.04 to 0.12.
Total Spin
The change in the molecular orbitals with varying c3 discussed in the previous section is reflected in the total and local spin values, which are plotted in Figures 4-7.

Total 〈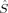 2〉 expectation values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)].
2〉 expectation values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)].

Partial 〈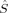 zO〉 values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)]. The atoms are defined by the Löwdin* partitioning scheme.
zO〉 values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)]. The atoms are defined by the Löwdin* partitioning scheme.

Partial 〈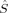 zFe〉 values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)]. The atoms are defined by the Löwdin* partitioning scheme.
zFe〉 values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)]. The atoms are defined by the Löwdin* partitioning scheme.

Partial 〈ŜFe1 · ŜFe2〉 values in a.u. for all spin state possibilities with varying c3 parameters [see eq. (16)]. The atoms are defined by the Löwdin* partitioning scheme.
For all spin states, the total 〈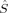 2〉 expectation values in Figure 4 increase with c3. For the S = 4, S = 3, and S = 2 determinants, the dependence on c3 is very weak. The values cover a range of 0.1 (S = 4, S = 3) or 0.2 (S = 2) atomic units, respectively. For the lower spin determinants based on standard initial guesses (Extended Hückel or hs MOs), with S = 1 and S = 0, all plots reflect the discontinuous behavior, which has been discussed earlier with respect to the change of the total energy. The dependence of 〈
2〉 expectation values in Figure 4 increase with c3. For the S = 4, S = 3, and S = 2 determinants, the dependence on c3 is very weak. The values cover a range of 0.1 (S = 4, S = 3) or 0.2 (S = 2) atomic units, respectively. For the lower spin determinants based on standard initial guesses (Extended Hückel or hs MOs), with S = 1 and S = 0, all plots reflect the discontinuous behavior, which has been discussed earlier with respect to the change of the total energy. The dependence of 〈 2〉 on c3 is much more pronounced for the lower spin than for the higher spin determinants. For the plot of the BS determinant 2(S0,EB/HB), whose continuity parallels the behavior of the same determinant in the total energy plot in Figure 3, the total 〈
2〉 on c3 is much more pronounced for the lower spin than for the higher spin determinants. For the plot of the BS determinant 2(S0,EB/HB), whose continuity parallels the behavior of the same determinant in the total energy plot in Figure 3, the total 〈 2〉 expectation value raises from 3.3 to 4.1 a.u. as c3 is increased. It can be concluded from eq. (2) that this indicates a gradually decreasing overlap of the magnetic orbitals of the 2(S0,EB/HB) determinant with increasing c3 parameter (provided a separation of the MOs into “closed-shell” orbitals (denoting pairs of α and β orbitals with an spatial overlap close to unity) and “magnetic” orbitals (which hardly overlap spatially with an orbital of the other spin quantum number) can be made as described earlier).
2〉 expectation value raises from 3.3 to 4.1 a.u. as c3 is increased. It can be concluded from eq. (2) that this indicates a gradually decreasing overlap of the magnetic orbitals of the 2(S0,EB/HB) determinant with increasing c3 parameter (provided a separation of the MOs into “closed-shell” orbitals (denoting pairs of α and β orbitals with an spatial overlap close to unity) and “magnetic” orbitals (which hardly overlap spatially with an orbital of the other spin quantum number) can be made as described earlier).
Local Spins
Figures 5 and 6 summarize the local 〈 zA〉 expectation values obtained with the Löwdin* partitioning scheme, which are equal to half the difference of the α and β electrons located on the atom A, for one of the Fe atoms (Fe1 in Fig. 1) and one of the bridging oxygen atoms (O1). The 〈
zA〉 expectation values obtained with the Löwdin* partitioning scheme, which are equal to half the difference of the α and β electrons located on the atom A, for one of the Fe atoms (Fe1 in Fig. 1) and one of the bridging oxygen atoms (O1). The 〈 z〉 fraction on all other atoms except the two iron centers and the bridging oxygen atoms is below a value of 0.1 atomic units.
z〉 fraction on all other atoms except the two iron centers and the bridging oxygen atoms is below a value of 0.1 atomic units.
The local spin analysis shows that there are approximately three unpaired electrons located at Fe1 for the hs (S = 4), the S = 3 and one for the S = 2 determinants [2(S2,H)]. The corresponding 〈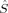 zFe1〉 values increase from 1.51 to 1.67, from 1.45 to 1.65, and from 1.33 to 1.66, respectively. On the other iron atom, which is not included in the plot, the local spins correspond to those given for B3LYP/structure 2 in Table 2. The remaining unpaired electrons are distributed mainly onto the oxygen bridges.
zFe1〉 values increase from 1.51 to 1.67, from 1.45 to 1.65, and from 1.33 to 1.66, respectively. On the other iron atom, which is not included in the plot, the local spins correspond to those given for B3LYP/structure 2 in Table 2. The remaining unpaired electrons are distributed mainly onto the oxygen bridges.
The spin delocalization from the metal centers onto the bridging oyxgen atoms is reduced for S = 4, S = 3 and 2(S2,H) as c3 increases. The 〈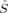 zA〉 values for these determinants show that there is a small sudden qualitative change in the MOs between c3 = 0.16 and c3 = 0.20. For S = 2, the two standard intitial guess MOs have converged to determinants with different local spin properties. The MOs based on an Extended Hückel guess have a 〈
zA〉 values for these determinants show that there is a small sudden qualitative change in the MOs between c3 = 0.16 and c3 = 0.20. For S = 2, the two standard intitial guess MOs have converged to determinants with different local spin properties. The MOs based on an Extended Hückel guess have a 〈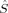 zFe1〉 value, which is quite constant around 0.8, and thus corresponds best to ideal local spins of 1 on both iron atoms. The MOs obtained starting from the hs MOs have most of the spin excess localized on the first iron atom, as indicated by the 〈
zFe1〉 value, which is quite constant around 0.8, and thus corresponds best to ideal local spins of 1 on both iron atoms. The MOs obtained starting from the hs MOs have most of the spin excess localized on the first iron atom, as indicated by the 〈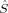 zFe1〉 value ranging from 1.32 to 1.66 (see also Table 2). This spin distribution is equal in energy to the symmetric one for c3 = 0.0, but gets more favored at larger c3.
zFe1〉 value ranging from 1.32 to 1.66 (see also Table 2). This spin distribution is equal in energy to the symmetric one for c3 = 0.0, but gets more favored at larger c3.
The S = 1 and S = 0 curves show the same discontinuities as the plots discussed in the preceding sections. The 2(S0,EB/HB) curve, which was constructed from the 2(S0,H,B3[0.04]) MOs as initial guess, has local 〈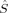 zFe1〉 values dropping from −1.36 to −1.68 a.u., which corresponds to an increase of local spin polarization with c3 as also observed for the higher spin determinants. On the other iron atom, which has not been plotted, the values are the same as for Fe1 with the signs inverted. This determinant is therefore the closest one can get to a BS determinant with a standard SCF guess for this MMO model complex. It effectively has three unpaired α electrons on one iron atom and three unpaired β electrons on the other one. The somewhat strange convergence behavior of the S = 1 guesses to solutions with simply interchanged iron spins, but energy differences of up to 40 kJ/mol, has already been adressed.
zFe1〉 values dropping from −1.36 to −1.68 a.u., which corresponds to an increase of local spin polarization with c3 as also observed for the higher spin determinants. On the other iron atom, which has not been plotted, the values are the same as for Fe1 with the signs inverted. This determinant is therefore the closest one can get to a BS determinant with a standard SCF guess for this MMO model complex. It effectively has three unpaired α electrons on one iron atom and three unpaired β electrons on the other one. The somewhat strange convergence behavior of the S = 1 guesses to solutions with simply interchanged iron spins, but energy differences of up to 40 kJ/mol, has already been adressed.
The 〈ŜFe1 · ŜFe2〉 expectation values can be employed for the calculation of the Heisenberg coupling constants according to eq. (7). Because only the hs and the BS determinant are needed for this calculation, only those two plots in Figure 7 shall be considered explicitely.
As far as the other curves are concerned, they display the already described qualitative discontinuities for the S = 1 and S = 0 determinants, but also additional ones for S = 2 and S = 3, which have not been observed at this c3 value in the other local spin, total spin, and energy plots. Such sudden qualitative changes of the orbitals that are not visible in the energy plots are also revealed by the plots of the local 〈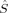 zFe1〉 and 〈
zFe1〉 and 〈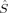 zO1〉 expectation values (for the S = 4 and S = 3 determinants in the former and the S = 3 and S = 2 determinants in the latter case). The important hs and BS 〈ŜFe1 · ŜFe2〉 curves [2(S4,E) and 2(S0,EB/HB)], however, are continuous. The hs values are close to the negative values of the BS case. The magnitude of both is increasing with c3. With values of maximal magnitude of 2.82 and −2.92, both are quite far form the values for ideal local spin eigenfunctions of 4 and −6.
zO1〉 expectation values (for the S = 4 and S = 3 determinants in the former and the S = 3 and S = 2 determinants in the latter case). The important hs and BS 〈ŜFe1 · ŜFe2〉 curves [2(S4,E) and 2(S0,EB/HB)], however, are continuous. The hs values are close to the negative values of the BS case. The magnitude of both is increasing with c3. With values of maximal magnitude of 2.82 and −2.92, both are quite far form the values for ideal local spin eigenfunctions of 4 and −6.
Dependence of Heisenberg Coupling Constants on the Exact Exchange Admixture
The absolute and relative energies of the different spin states depend strongly on the exact exchange admixture in the modified B3LYP density functional, as illustrated earlier. Because the Heisenberg coupling constant JFe1Fe2 is a function of the energy difference between the hs and the BS determinant, a dependence of JFe1Fe2 on the exact exchange admixture is also to be expected. The dependence of the energy splitting is displayed in Figure 8. It is supplemented by a plot of the difference of the 〈ŜFe1 · ŜFe2〉 expectation value for the hs and the BS determinant, which is needed for the calculation of coupling constants with Clark and Davidson's local spin values.

Differences between hs and BS total energies in kJ/mol and 〈ŜFe1 · SFe1〉 expectation values in a.u. with varying c3 parameters.
Whereas both curves in Figure 8 decrease as the exact exchange increases, the energy difference depends much more on the c3 parameter than the local spin difference. In accordance with the findings for mononuclear transition metal complexes, the hs state is more favored by an increasing exact exchange admixture. The curvature of the energy difference plot can be traced back to the energy plots in Figure 3 discussed earlier: whereas the hs plot is bent upwards, the BS coupling constant behaves almost linear with c3. Heisenberg coupling constants JFe1Fe2 calculated with Clark and Davidson's local spin expectation values,31, 50 with Noodleman's35 and with Yamaguchi's19 expression, are plotted in Figure 9 as a function of the exact exchange admixture parameter c3.

Heisenberg coupling constants JFe1Fe2 in cm−1 as a function of the exact exchange admixture c3 in a modified B3LYP functional.
The Heisenberg coupling constants JFe1Fe2 calculated with the expressions of Noodleman eq. (4) or Yamaguchi eq. (6) become less negative as c3 is increased. JFe1Fe2 is proportional to the negative energy difference between the hs and the BS determinant in all three expressions, and the denominator is independent of c3. Therefore, this behavior of JFe1Fe2 can be expected from the decline of the energy difference with increasing c3 displayed in Figure 8. Both curves agree very well quantitatively, which is in accord with Yamaguchi's statement that his expression approaches Noodleman's in the limit of weakly overlapping magnetic orbitals. According to the conclusion drawn earlier, that the magnetic orbital overlap decreases with increasing c3, the differences between the results obtained with both expressions nearly vanish as c3 gets larger.
The coupling constants calculated with Clark and Davidson's local spins from eq. (7), however, become nearly constant with an increasing value of c3, starting from c3 = 0.15. This is possible, because the variation of the energy difference can be compensated by the variation of the 〈ŜFe1 · ŜFe2〉 expectation value (see Fig. 8). In the region below c3 = 0.15, the difference between the Clark and Davidson's and Noodleman's/Yamaguchi's values is quite constant, the former being about 100 cm−1 more negative than the latter. This large difference can be explained by the fact that the hs and the BS determinant are not eigenfunctions of the local spin operators 〈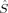 〉 and 〈
〉 and 〈 〉 with the same eigenvalues (see earlier). The JFe1Fe2 values calculated with the BP86 functional for structures 2 with the three different methods of Clark and Davidson, Noodleman and Yamaguchi are −494, −260 and −271 cm−1, respectively. These data are in the range of the JFe1Fe2 coupling constant of −376 cm−1 obtained by Lovell, Noodleman et al.76 for an extended version of Siegbahn's model investigated here. This extended model contains additional second- and third-shell amino acid residues and two water molecules. The authors claim the result to be “in reasonable accord, both in sign and in magnitude, with the estimation from Mössbauer spectroscopy (J < −30 cm−1).” As far as the qualitative agreement of calculated and experimental data is concerned, the result is in both cases that the antiferromagnetically coupled state with S = 0 is the ground state of the intermediate Q of MMOH. From a quantitative point of view, however, a difference by the factor of ten is not satisfactory. This difference might be either due to the method chosen for the calculation of the Heisenberg coupling constant, to an inappropriate model cluster, or to a breakdown of the Heisenberg model, provided the possibility of experimental inaccuracies is not taken into account.
〉 with the same eigenvalues (see earlier). The JFe1Fe2 values calculated with the BP86 functional for structures 2 with the three different methods of Clark and Davidson, Noodleman and Yamaguchi are −494, −260 and −271 cm−1, respectively. These data are in the range of the JFe1Fe2 coupling constant of −376 cm−1 obtained by Lovell, Noodleman et al.76 for an extended version of Siegbahn's model investigated here. This extended model contains additional second- and third-shell amino acid residues and two water molecules. The authors claim the result to be “in reasonable accord, both in sign and in magnitude, with the estimation from Mössbauer spectroscopy (J < −30 cm−1).” As far as the qualitative agreement of calculated and experimental data is concerned, the result is in both cases that the antiferromagnetically coupled state with S = 0 is the ground state of the intermediate Q of MMOH. From a quantitative point of view, however, a difference by the factor of ten is not satisfactory. This difference might be either due to the method chosen for the calculation of the Heisenberg coupling constant, to an inappropriate model cluster, or to a breakdown of the Heisenberg model, provided the possibility of experimental inaccuracies is not taken into account.
Conclusion
Within the KS density functional theory, we have systematically investigated the dependence on the functional of total energies, of local and total spin expectation values, and of Heisenberg coupling constants calculated according to three different “recipes.” For this purpose, two standard density functionals, namely the pure functional BP86 and the hybrid functional B3LYP, have been chosen, as these two comparatively old representatives of pure and hybrid functionals are still reliable because they could not yet be substituted by new functionals of higher accuracy. However, it is now well recognized that the exact exchange admixture c3, which distinguishes the hybrid from the pure functionals, becomes decisive for spin states of transition metal complexes.15 Consequently, a modified B3LYP functional with an exact exchange admixture c3 varying from zero to 24% is included in this study. To gain qualitative insight into the electronic structure of the optimized states, we have compared Clark and Davidson's local spin expectation values obtained with modified Löwdin projectors to ideal local spin values for the different functionals. Two different (unspecific) initial guesses for the MOs have been chosen on purpose to screen a sufficiently large portion of the parameter space for the total energy to ensure that the SCF procedure may converge to different stationary points.
-
The total energy difference measured with respect to the well-defined RKS reference determinant becomes more negative when c3 is increasing for all spin states under investigation so that the energies are spread over a larger range when c3 is higher. When c3 is large (say higher than 12%), the hs and the BS determinant are the two states with lowest energy with the BS state being the ground state. For lower c3 values, energies of intermediate spin states are lying between both. The higher the exact exchange admixture, the more the hs determinant is favored, so that the energy differences between the hs and BS determinants decreases with increasing c3.
-
It is also observed that both the local spin polarization and the spin contamination increase with c3. The same behavior of the spin contamination was found by Rode and Werner for dicopper complexes,77 whereas for mononuclear complexes, the spin contamination turned out to decrease with an increasing exact exchange admixture.13
-
The coupling constant JFe1Fe2 for the MMOH model complex varies between −129 and −494 cm−1, depending on the exact exchange admixture and the method with which JFe1Fe2 is calculated. These results confirm that the antiferromagnetically coupled state with S = 0 is the ground state of the intermediate Q of MMOH.
-
The high degree of spin delocalization onto the bridging oxygen atoms and the fact that neither the hs nor the BS determinant are eigenfunctions of the local spin operators raise doubts whether this system can be properly described by the Heisenberg model. However, it may be anticipated that the variation of the coupling constants will also be large in cases suitable for a phenomenological Heisenberg Hamiltonian (see also the work of Yamaguchi et al.19-21). However, in view of the fact that the coupling constant is a derived quantity subject to various assumptions in experiment and theory, we shall not follow Yamaguchi and collaborators and advocate for a specific functional for the calculation of these parameters.
-
With respect to the standard MO guesses applied in this work, we note that the convergence behavior of both Extended Hückel and hs guess MOs gets the more difficult to control the lower the spin state is. For the hs (S = 4) and the septet (S = 3) state, both always converge to chemically reasonable solutions. As far as the lower spin states are concerned, the convergence is more difficult to control. Whereas at low exact exchange admixtures of zero or 4%, both initial guesses with S = 0 converge to the BS solution, for higher c3 values they do not. In these cases, the MOs of a converged BS determinant need to be used as initial guess.
Altogether, the data presented, especially the strong variations of JFe1Fe2, indicate that the reliable calculation of coupling constants by means of density functional theory is still unsettled. The data show, however, that modifications of the exchange part in the density functional as well as a detailed investigation on how much deviations from local spin eigenfunctions affect the validity of the Heisenberg model and the comparability to experimental results. Based on the systematic study presented here, we may recommend to check DFT calculations for polynuclear clusters for their stability with respect to exact exchange admixture rather than to test many different functionals.
It would be desirable to have a tool at hand that circumvents the tedious manual setup of the MO initial guess described in ref.34 and that solves the convergence problems of standard initial guess methods. A scheme that controls SCF convergence in each iteration step and that allows one to start from any initial guess is currently investigated in our laboratory.
Acknowledgements
The authors gratefully acknowledge discussions with Professor W. Boland on desaturase activity, which stimulated this work.
Appendix A: SCF Convergence
To which global or local minimum solution the SCF algorithm converges, depends strongly on the initial guess of the molecular orbitals, and is usually not explicitly controlled in the iterations. In KS-DFT calculations, it furthermore depends on the chosen density functional. Both parameters are fixed at the beginning of a SCF iteration. The construction of an initial guess that converges to a Slater determinant with certain local spin properties can be an art in itself, which is described in detail in ref.34. In this work, the SCF convergence of initial guess MOs which are comparatively easy to construct shall be briefly investigated. These are MOs obtained from Extended Hückel theory on the one hand, and the converged KS-DFT MOs of the hs (S = 4) complex (obtained for the corresponding functional and molecular structure) on the other hand.
The question that shall be investigated here is to which stationary points standard initial guesses converge. In other words, we shall check in this appendix the stability of SCF convergence for open-shell UKS determinants. Furthermore, we are interested in sampling a rather large part of the energy hypersurface in the parameter space of the MO coefficients to obtain other potential local minima. The influence of the density functional on the convergence of a given initial guess is assessed by comparing the converged MOs obtained with the modified B3LYP density functional with different c3 parameters.
For mononuclear transition metal complexes, the question to which local spin state an initial guess converges is usually answered by the chosen total spin. If there are no open-shell ligands and if spin delocalization is negligible, the excess spin will be located on the metal atom, and the metal local spin will therefore be equal to the total spin. The problem with polynuclear clusters is that there are usually several possibilites to obtain a given total spin by combinations of local spins (see, e.g., Table 3 and the discussion in an earlier section). The focus in this appendix will therefore be on a comparison of both initial guesses.
It turns out that the Extended Hückel guess tends to converge to symmetric and only slightly spin-polarized solutions. For example, the hs guesses for the quintet (S = 2) converge to solutions corresponding to local iron spins of 2 and 0 for all c3 parameters, whereas the Extended Hückel guesses converge to solutions with local spins of 1 on both iron atoms. The dependence of the local spins on one of the iron atoms, Fe1, is depicted in Figure 6. The local spins on the other iron atoms have not been included, but the reader may be referred to the corresponding values for Fe2 in Table 2.
The tendency of the Extended Hückel guess to favor less spin-polarized solutions can be rationalized by the fact that the Extended Hückel guess distributes the excess spin over the whole molecule, which results in very low local spins on the iron atoms and on the oxygen bridge, as can be understood from Table 4.
| Spin state | Structure | 〈 2〉 2〉 |
 zFe1 zFe1 |
 zFe2 zFe2 |
 zO1 zO1 |
 zO2 zO2 |
〈 Fe1· Fe1· Fe2〉 Fe2〉 |
|---|---|---|---|---|---|---|---|
| S4 | 1 | 20.02 | 0.13 | 0.13 | 0.07 | 0.08 | −0.02 |
| 2 | 20.02 | 0.13 | 0.15 | 0.10 | 0.12 | −0.01 | |
| S3 | 1 | 12.02 | 0.07 | 0.07 | 0.05 | 0.04 | −0.05 |
| 2 | 12.02 | 0.08 | 0.06 | 0.06 | 0.05 | −0.04 | |
| S2 | 1 | 6.03 | 0.04 | 0.04 | 0.003 | 0.003 | −0.04 |
| 2 | 6.03 | 0.04 | 0.03 | 0.003 | 0.003 | −0.04 | |
| S1 | 1 | 2.03 | 0.03 | 0.03 | 0.002 | 0.002 | −0.04 |
| 2 | 2.03 | 0.04 | 0.03 | 0.002 | 0.002 | −0.04 |
- The S = 0 results have not been included because all values are equal to zero.
The total spins in the first column indicate that the spin contamination is low, as can be expected from eq. (2) for determinants with negligible local spin polarization. The very small local  zA values, which are as low as 0.13 for the iron atoms in the hs determinant, where values close to 2 are expected and have been found for a converged determinant, together with the local spin on the atoms not included in Table 4, which are in the same range, indicate that the spin is distributed over the whole molecule. Nonetheless, it is astonishing that the Extended Hückel guess converges to chemically reasonable determinants at all for polynuclear complexes.
zA values, which are as low as 0.13 for the iron atoms in the hs determinant, where values close to 2 are expected and have been found for a converged determinant, together with the local spin on the atoms not included in Table 4, which are in the same range, indicate that the spin is distributed over the whole molecule. Nonetheless, it is astonishing that the Extended Hückel guess converges to chemically reasonable determinants at all for polynuclear complexes.
It is furthermore remarkable that the discontinuities of both guesses with respect to the c3 dependence of the expectation values of the corresponding converged determinants occur at the same points, even if both converge to different solutions (see, e.g., the triplet plots in Fig. 6). For the higher spin states (S = 3 and S = 4), however, both guesses always converge to the same solution, because there is not much choice (if the interchange of local spins of 2 and 1 for S = 3 is excluded). Interestingly, for S = 0, both guesses also always converge to the same determinant, regardless of c3. For low exact exchange admixtures (0 to 4%), the BS determinant is obtained, whereas for higher c3 values, the guesses converge to chemically inrelevant high-energy solutions. When the BS MOs obtained with c3 = 0.04 are employed, however, the BS solution is obtained for the higher c3 values as well.

