

Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells
Abstract
Recent studies in acute myeloid leukemia (AML) suggest activation of pro-proliferative signaling cascades including those mediated by protein kinase C (PKC) represent a poor prognostic factor for patients. The classical PKC isoforms α and β generally support survival signaling and have emerged as important targets for anti-cancer therapy. Enzastaurin is a PKC β inhibitor and is in clinical trials for lymphomas, gliomas, and lung cancer. Presently, it is not known if enzastaurin could be effective against AML. In the current study, we found that high dose enzastaurin was found to promote apoptosis in the AML-derived cell lines and in blast cells from AML patients. The mechanism of cell death, however, likely does not involve PKC β as another PKC β inhibitor was not toxic to AML cell lines and did not promote enzastaurin-induced cell killing. While enzastaurin is fairly specific for PKC β, the agent can inhibit other PKC isoforms at higher concentrations. Enzastaurin was effective at inhibiting PKC α phosphorylation and membrane localization in the AML cell lines and suppressed phosphorylation of BCL2. Furthermore, enzastaurin suppressed activation of ERK (which can be activated by PKC α). Analysis of the serine/threonine phosphorylation profile in HL60 cells after enzastaurin treatment revealed that the drug inhibits the phosphorylation of a distinct set of proteins while promoting phosphorylation of another set of proteins. This suggests the drug may regulate multiple signaling pathways. Taken together, these findings suggest that enzastaurin could be effective in the therapy of AML. J. Cell. Biochem. 112: 1696–1707, 2011. © 2011 Wiley-Liss, Inc.
The protein kinase C (PKC) family of serine/threonine kinases consists of 12 known members found in three groups based on structure and activation criteria [Corbalán-García and Gómez-Fernández, 2006; Griner and Kazanietz, 2007; Redig and Platanias, 2007, 2008; Newton, 2010]. The classical PKC family (cPKC) includes α, βI, βII, and γ. The cPKC isoforms require calcium and are activated by diacylglycerol (DAG). The novel PKC (nPKC) isoforms are activated by DAG like the cPKC family, but these kinases do not require calcium. The nPKC family includes δ, ε, η, σ, and θ. Finally, the atypical PKC (aPKC) family does not require calcium and is not activated by DAG. The aPKC family includes ι, λ, and ζ. The cPKC and nPKC family members have two PKC-1 homology domains (C1A and C1B) that are responsible for binding DAG [Corbalán-García and Gómez-Fernández, 2006; Griner and Kazanietz, 2007]. Among cancer biologists there has been great interest in the cPKC and nPKC family members as these kinases are activated by potent carcinogens [e.g., phorbol esters; Griner and Kazanietz, 2007]. Phorbol esters are believed to promote tumorigenesis by binding the C1 domains to activate pro-proliferative PKC signaling [Castagna et al., 1982; Corbalán-García and Gómez-Fernández, 2006]. The sphingolipid ceramide appears to bind the single C1 domain in the aPKC members which may activate signaling cascades that promote growth arrest or death [Müller et al., 1995; Wang et al., 2005]. While ceramide may activate atypical PKC family members, the sphingolipid is effective at inhibiting classical PKC isoforms by promoting dephosphorylation of the kinase possibly via PP1 (Kitatani et al., 2007).
The role of the various PKC isoforms in tumorigenesis is complex. For example, PKC α might act as a tumor suppressor in one cell type [e.g., pancreatic and mammary; Detjen et al., 2000; Griner and Kazanietz, 2007] but acts as a supporter of malignant cell growth in another cell type [e.g., lymphoid and myeloid cells; Jiffar et al., 2004; Kornblau et al., 2006; Kurinna et al., 2006]. The PKC α and PKC β isoforms generally support pro-survival signaling in hematopoitic cells [Redig and Platanias, 2008]. Thus, it is no surprise that the PKC α and PKC β isoforms have been implicated in tumorigenesis and the support of chemoresistance in leukemia [Redig and Platanias, 2008]. PKC α has been shown to phosphorylate BCL2 in acute leukemia cells resulting in greater anti-apoptotic function [Ruvolo et al., 1998; Jiffar et al., 2004]. Activation of multiple signaling pathways including PKC α has been shown to be a poor prognostic factor in acute myeloid leukemia [AML; Kornblau et al., 2006]. PKC βII is critical for BCR-ABL-mediated leukemogenesis [Perrotti et al., 2000; Xenaki et al., 2004]. Introduction of PKC βII into hematopoietic progenitor cells promotes cell survival during cytokine (i.e., IL-3) withdrawal. PKC βII, however, does not block chemotherapy drug-induced apoptosis, at least when araC was the chemotherapeutic agent [Xenaki et al., 2004].
The potential roles for PKC α and PKC β in supporting cancer cell growth have inspired a number of strategies to target these kinases for the treatment of a number of malignancies [Mackay and Twelves, 2007]. Staurosporine was identified as a PKC inhibitor which targets the conserved ATP-binding domain but the drug inhibits many other kinases [Mackay and Twelves 2007]. Staurosporine derivatives such as PKC412 and UCN01 have been developed with improved specificity and are in use in clinical trials for solid tumors and hematologic malignancies [Propper et al., 2001; Sausville et al., 2001; Sampath et al., 2006; Mackay and Twelves, 2007] The Lilly compound enzastaurin (LY317615) was found to be fairly specific for PKC β and was originally developed as an anti-angiogenesis agent based on the role of PKC β in that process [Teicher et al., 2002a; Graff et al., 2005; Ma and Rosen, 2007]. At higher concentrations, the drug can inhibit other PKC isoforms including PKC α [Teicher et al., 2002a]. Enzastaurin has shown promise in a recent Phase II clinical trial for diffuse large B-cell lymphoma [Robertson et al., 2007]. Since lung cancer cells exhibit elevated expression of both PKC α and PKC β, and enzastaurin can effectively inhibit both kinases at concentrations used in therapy, the drug is being used in clinical trials for lung cancer [Herbst et al., 2007]. Considering that PKC plays a key role in leukemia cell survival [Redig and Platanias, 2008], a strategy using enzastaurin for the therapy of AML is appealing. A recent study has shown that enzastaurin promotes death ligand-induced apoptosis in leukemia cells [Meng et al., 2010]. However, at present it is unclear how enzastaurin affects AML cells. In the present study, we have found that PKC α and PKC β gene expression levels are significantly elevated in blast cells derived from AML patients compared to counterpart CD34+ cells derived from normal donors. Enzastaurin was found to be effective at promoting apoptosis in AML cell lines and leukemic blast cells from AML patients. While suppression of PKC β does not appear to be required for enzastaurin-induced cell death of the AML cells, the drug was effective at suppressing PKC α phosphorylation and membrane localization, inhibiting ERK phosphorylation, blocking BCL2 phosphorylation, and altering phosphorylation status of a distinct set of proteins. These results suggest that enzastaurin may be an effective agent in the therapy of AML.
MATERIALS AND METHODS
Materials
All reagents used were purchased from commercial sources unless otherwise stated. Enzastaurin was kindly provided by Eli Lilly and Company (Indianapolis, IN) or purchased from LC Laboratories (Woburn, MA).
Cell Lines and Patient Samples
OCI-AML2 and OCI-AML3 cells were kindly provided by M. D. Minden (Ontario Cancer Institute, Toronto, ON, Canada). U937, KG1, K562, THP-1, and HL60 cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were maintained in RPMI medium 1640 supplemented with 5% fetal bovine serum and 5% calf serum at 37°C in 5% CO2. Samples of peripheral blood were obtained for in vitro studies from patients with newly diagnosed or recurrent AML with high (more than 40%) blast count. Informed consent was obtained following institutional guidelines. Clinical data for AML patient samples used in the in vitro assays are presented in Table I. Patient karyotypes were obtained by conventional methods as part of the standard diagnostic workup, typically on samples derived from bone marrow. Samples were obtained from all patients prior to treatment. Mononuclear cells were isolated by centrifugation through Ficoll-Hypaque (Sigma Chemical Co, St. Louis, MO) density gradients. Samples from healthy bone marrow donors were selected for CD34+ cells using a Mini-Macs magnetic-antibody separation column (Miltenyi Biotech, Auburn, CA) as directed by the manufacturer.
| Patient number | FAB | WBC | PB blast | BM blast | Cytogenetics |
|---|---|---|---|---|---|
| 1 | M4 | 42.1 | 84 | 84 | Hyperdiploid clone 47,XY, + 8[14],Diploid male karyotype 46,XY[6] |
| 2 | M1 | 23.6 | 63 | 25 | Diploid male karyotype 46,XY[20] |
| 3 | M5 | 0.7 | 5.4 | 84 | Pseudodiploid clone 46,XY,t(9;11)(p22;q23)[20] 44,X,del(X)(q26q28),-5,del(9)(q32q34),-13,-18, + del(20)(q11.2q13.3)- 22, + mar[2],44,sl,del(17)(q23q25)[10],Hypodiploid metaphase 44, |
| 4 | M1 | 9.9 | 96 | NA | sl, + der(1;19)(q10;p10),-18[1] Pseudodiploid clone 46,XX,del(12)(p11.2p13)[3],46,XX, del(12)(p11.2p13),inv(12)(p13q22)[16],Hyperdiploid metaphase 47, |
| 5 | M4 | 16.5 | 48 | 62 | XX, + 8,del(12)(p11.2p13),inv(12)(p13q22)[1] |
| 6 | UNK | 11.3 | 98 | 90 | Pseudodiploid clone 46,X,t(X;3)(p22.1;p21),t(3;11)(p21;p13)[20] |
| 7 | M2 | 9.4 | 80 | 73 | 10/30/2007:Diploid female karyotype 46,XX[20] 12/3/2007:46,XX,t(5;19)(q15;q13.3)[15],46,XX t(5;19)(q15;q13.3)[cp4],46,XX,del(4)(q21q27),t(5;13)(q15;q32), |
| 8 | M2 | 16.3 | 98 | 97 | add(19)(q13.4)[1] |
| 9 | M4 | 0.5 | 60 | 78 | 11/30/2007:Diploid female karyotype 46,XX[20] |
- FAB, French-American-British; WBC, white blood count; PB, peripheral blood; BM, bone marrow.
Gene Expression Analysis
Leukemia-derived cell lines (OCI-AML3, U937, KG1, K562, HL60), blast cells from 75 AML patients, and CD34+ cells from six normal donors were washed, lysed with RNA Stat 60 (Tel-Test, Friendswood, TX), and then stored at −80°C pending final extraction of total RNA. To ensure complete removal of trace genomic DNA or other factors that could interfere with downstream enzymatic processes (i.e., heparin anticoagulant), all RNA samples were subjected to final purification using RNeasy Mini Columns (Qiagen, Valencia, CA) with on-column treatment by DNAse I as directed by the manufacturer. We prepared cDNA from 1.0 µg of total RNA per 20 µl mix containing 0.07 µg/µl random-sequence hexamer primers, 1 mM dNTPs, 5 mM DTT, 0.2 U/µl SuperAsin RNAse inhibitor (Ambion, Austin, TX), and 10 U/µl SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA). RNA and primers were denatured 5 min at 70°C and then chilled on ice. All components except enzyme were added and the mixture was incubated at room temperature for 2 min to allow nucleic acids to anneal. After addition of reverse transcriptase, the mixture was incubated for 10 min at 25°C, then 1 h at 50°C, followed by heat-inactivation of the enzyme for 15 min at 72°C. All cDNAs were stored at −80°C when not in use. To verify the complete removal of any residual genomic material in the real-time PCR assays, we incubated in parallel 1.0 µg of total RNA per 20 µl of a mix containing all components except reverse transcriptase.
We carried out real-time PCR using an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). We ran duplicate 25 µl reactions containing 0.5 µl cDNA; reactions were repeated if the Ct values were more than 0.25 cycles apart. As primers and probes we used TaqMan Gene Expression Assays (Applied Biosystems) specific for the genes of interest in this study (Table II) as directed by the manufacturer. We used ABL and b2M as housekeeping genes to normalize gene expression. To calculate the relative abundance (RA) of each transcript of interest relative to that of housekeeping gene, the following formula was employed: 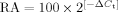 , where ΔCt is the mean Ct of the transcript of interest minus the mean Ct of the transcript for ABL or b2M. The Mann–Whitney Rank Sum Test was used to test for a difference in gene expression of PKC isoforms between normal CD34+ bone marrow cells and blast cells from AML patients. Differences were considered statistically significant when P < 0.05. Statistical analysis was performed with Sigma Stat computer software (SSPS, Chicago, IL).
, where ΔCt is the mean Ct of the transcript of interest minus the mean Ct of the transcript for ABL or b2M. The Mann–Whitney Rank Sum Test was used to test for a difference in gene expression of PKC isoforms between normal CD34+ bone marrow cells and blast cells from AML patients. Differences were considered statistically significant when P < 0.05. Statistical analysis was performed with Sigma Stat computer software (SSPS, Chicago, IL).
| Common name | Symbol | Chromosome location | ABI assay ID |
|---|---|---|---|
| PKC α | PRKCA | 17q22-q23.2 | Hs00176973_m1 |
| PKC β | PPP2R1B | 16p11.2 | Hs00176998_m1 |
| PKC δ | PPP2R2A | 3p21.31 | Hs00178914_m1 |
| PKC ε | PPP2R5A | 2p21 | Hs00178455_m1 |
| β-2-microglobulin | B2M | 15q21-q22.2 | Hs99999907_m1 |
| ABL1 | ABL1 | 9q34.1 | Hs00245445_m1 |
Analysis of Cell Viability and Apoptosis
Cells were treated with various doses of enzastaurin for times up to 72 h in media containing 1% fetal bovine serum. Where appropriate, cells were pretreated for 1 h prior to enzastaurin addition with 40 µM caspase 3 inhibitor (Z-DEVD-FMK; Calbiochem, La Jolla, CA) or 10 nM okadaic acid (Calbiochem). Cells were also treated alone or co-treated with 200 nM PKC β inhibitor (Calbiochem). Cell viability was measured by trypan blue dye exclusion assay. To determine that the mechanism of cell death was apoptosis, cells were stained with Annexin V/TMRM (tetramethyl rhodamine methyl ester) and the percentages of apoptotic cells were assessed by flow cytometry. Cells were washed in PBS, resuspended in binding buffer containing Annexin V (Roche Diagnostics, Indianapolis, IN). Apoptotic cells were identified as positive for Annexin V staining using a Becton Dickinson LSR II flow cytometer (Becton Dickinson, San Jose, CA). Differences in cell viability and percent apoptosis in the cell lines were considered statistically significant when P < 0.05 using Student's t-test.
Metabolic Labeling, Immunoprecipitation, and Immunoblotting Analysis
OCI-AML3 cells were treated with 5 µM enzastaurin for 3 h during metabolic labeling with 32P-orthophosphate and BCL2 phosphorylation status was analyzed by immunoprecipitation as previously described [Ruvolo et al., 1998]. Anti-sera from Santa Cruz Biotechnology (Santa Cruz, CA) was used to immunoprecipitate BCL2. Immunoprecipitated protein was electrophoresed in a 12% acrylamide/0.1% SDS gel, transferred to nitrocellulose, and exposed to Kodak X-Omat film at −80°C. To confirm the identity of phosphorylated bands as BCL2, the same blot was used for Western blotting with an antibody that was different than the one used for immunoprecipitation. Anti-BCL2 anti-sera from Dako (Carpinteria, CA) was used in the Western analysis.
Western Blot Analysis
HL60 cells, OCI-AML3 cells, or blast cells from AML patients were sonicated in lysis buffer [62.5 mM Tris (pH 8.0), 2% SDS, 10% glycerol, 100 µM AEBSF, 80 nM Aprotinin, 5 µM Bestatin, 1.5 µM E-64, 2 µM leupeptin, 1 µM Pepstatin, 500 µM sodium orthovanadate, 500 µM glycerol phosphate, 500 µM sodium pyrophosphate, and 50 µM DTT], and protein (5 × 105 cell equivalents) was subjected to electrophoresis using 10–14% acrylamide/0.1% SDS gels. Proteins were transferred to a nitrocellulose membrane and Western blotting analysis was performed with antibodies against phospho-PKC α (Cell Signaling, Beverly, MA), PKC α (Cell Signaling), phospho-PKC β (Cell Signaling), PKC β (Cell Signaling), phospho-ERK (Cell Signaling), ERK (Cell Signaling), phospho-serine 70 BCL2 (Cell Signaling), BCL2 (Dako) Actin (Sigma, St. Louis, MO), and Tubulin (Sigma). Western blots were developed using an ECL kit (Amersham, Piscataway, NJ).
Two-dimensional Electrophoresis
Two-dimensional electrophoresis was performed by Kendrick Labs, Inc (Madison, WI). Isoelectric focusing was carried out using 2% ampholines of pH 3.5–pH 10 (LKB/Pharmacia, Baltimore, MD) for 9,600 V/h in a tube gel. Each tube gel was then electrophoresed using a 10% acrylamide SDS slab gel for 5 h at 25 mA/gel. Duplicate gels were obtained from each sample. Proteins were transferred to nitrocellulose and Western analysis was performed with phospho-ser/phospho-thr antibodies from Qiagen. Films were scanned with a laser densitometer (Model PDSI; Molecular Dynamics Inc, Sunnyvale, CA). The images were analyzed using Progenesis PG240 software with TT900 (version 2006; Nonlinear Dynamics). Student's t-test values were generated by Progenesis software for an n of 4 films.
Immunofluorescence (IF) Microscopy
OCI-AML3 cells were treated with vehicle (0.1% DMSO) or with 1 µM or 10 µM enzastaurin for 24 h. Cells were spun onto slides using a Statspin Cytofuge 12 (Cytofuge, Westwood, MA), fixed with 4% paraformaldehyde and permeablized with 100% methanol. Cells were incubated in blocking solution (3% FBS/1% BSA/1× PBS). PKC α or PKC β rabbit polyclonal antibodies (both from Santa Cruz) were added to cells at 1:200 dilution in blocking solution. Cells were washed and secondary antibody (Alexa 488 tagged donkey anti-rabbit; Invitrogen, Eugene, OR) was added in blocking solution. Cells were washed, stained with DAPI, washed again, and visualized using an Olympus FV500 Laser Scanning Confocal microscope (Center Valley, PA).
RESULTS
Gene Expression of PKC β is Elevated in AML Blast Cells Compared to Normal CD34+ Cells.
We measured gene expression of PKC α, PKC β, PKC δ, PKC ε by RT-PCR in a number of myeloid leukemia cell lines including OCI-AML3, U937, KG1, K562, and HL60. A common feature among the cell lines was that for each cell line PKC β and PKC δ were the more highly transcribed isoforms and little if any PKC ε was detected in the myeloid cell lines (Fig. 1A). As shown in Figure 1A, gene expression of PKC α varied among the various myeloid leukemia cell lines. We next surveyed the relative abundance of transcripts encoding the various PKC isoforms among 65 patients newly diagnosed with AML using real time PCR. We included assays for PKC α, PKC β, PKC δ, and PKC ε (Table II). Average expression of the chosen PKC isoforms in the samples from 65 AML patient and six bone-marrow transplant donors is depicted in Figure 1B. There was a significant increase in the expression of PKC α (P = 0.027), PKC β (P = 0.0002), and PKC δ (P < 0.0001) when comparing AML blast cells and normal BM cells. This finding suggests these kinases may play a role in AML biology.

PKC β and PKC δ expression is generally elevated IN AML blast cells compared to normal CD34+ bone marrow cells. Real-time PCR was performed as described in Materials and Methods section. Expression of PRKCA (PKC α), PRKCB (PKC β), PRKCD (PKC δ), and PRKCE (PKC ε) genes are presented as relative to 100 copies of b2M for AML-derived cell lines (A) and as relative to 100 copies of ABL1for CD34+ cells derived from bone marrow from normal donors (N = 6) and blast cells derived from AML patients (N = 75; B). P values were derived as described in Materials and Methods section. Statistically significant differences from expression in normal bone marrow cells (P < 0.05) are marked by “*.”
Enzastaurin Promotes Apoptosis in Human AML-Derived Cell Lines
To investigate the use of enzastaurin as a cytotoxic agent against AML cells, OCI-AML3 were used in a dose response study. While enzastaurin inhibits PKC β in the nanomolar range, it is in the low micromolar range where the drug has demonstrated effectiveness against a wide variety of cancer cell lines including leukemia cells [Graff et al., 2005; Meng et al., 2010]. OCI-AML3 cells were treated with vehicle (0.1% DMSO), 1, 5, or 10 µM enzastaurin for 24, 48, and 72 h. Cell viability was assessed by trypan blue exclusion. As shown in Figure 2A, enzastaurin suppressed cell growth of cells but only at higher doses of the drug (i.e., 5 or 10 µM enzastaurin). Similar patterns of cell growth inhibition were observed with OCI-AML2 and THP-1 cells (data not shown). To determine if enzastaurin promotes apoptosis, HL60 and OCI-AML3 were treated with 5 µM enzastaurin for 24, 48, and 72 h. Cell viability was assessed by trypan blue exclusion. As shown in Figure 2B, enzastaurin potently killed HL60 cells but was less effective against the OCI-AML3 cells. While roughly 50% of HL60 cells were killed after 72 h with 5 µM enzastaurin only ∼26% of OCI-AML3 cells were killed by the drug under those conditions (Fig. 2B). Cell death induced by enzastaurin was significant as compared to vehicle control (i.e., DMSO treated) in all cases for both cell lines (P < 0.006). To determine if an apoptotic mechanism was involved, induction of apoptosis in enzastaurin-treated cells was observed by identifying Annexin V positive cells (which indicates exposure of plasma membrane phosphatidyl serine). Enzastaurin (5 µM) promoted Annexin V staining of HL60 cells and to a lesser degree OCI-AML3 cells. As shown in Figure 2C, flow cytometry analysis of untreated HL60 cells and cells treated with 5 µM enzastaurin for 72 h indicate that nearly one-third of cells were Annexin V positive after staining. After 72-h treatment of OCI-AML3 cells with 5 µM enzastaurin, only ∼18% were apoptotic. These data indicate that enzastaurin has varying effects on the promotion of apoptosis in AML cell lines. Next we investigated whether enzastaurin-induced apoptosis involves a caspase-dependent mechanism. Apoptosis assay measuring Annexin V positive cells revealed that pretreatment of HL60 cells with 40 µM caspase-3 inhibitor partially protected cells from treatment with 5 µM enzastaurin after 72 h (Fig. 2D). This result suggests that the mechanism of enzastaurin-induced cells death involves caspase-3 but since protection with the caspase inhibitor was not complete, the process may require additional mechanisms.

Effect of enzastaurin on cell viability and caspase-dependent apoptosis in two AML cell lines. A: OCI-AML3 cells were treated with vehicle (0.1% DMSO; marked 0), 1 µM enzastaurin, 5 µM enzastaurin, or 10 µM enzastaurin for 24, 48, or 72 h. Cell death was assessed by trypan blue exclusion assay. B: HL60 cells and OCI-AML3 cells were untreated or treated with 10 µM enzastaurin for 24, 48, or 72 h. Cell death was assessed by trypan blue exclusion assay. Error bars represent the mean ± SD from three independent experiments. Statistically significant differences from cell viability in DMSO-treated cells (standard t-test; P < 0.05) are marked by “*.” C: HL60 cells and OCI-AML3 cells were untreated or treated with 10 µM enzastaurin for 48 h. Cell apoptosis was assessed by flow cytometry after Annexin V staining. Error bars represent the mean ± SD from three independent experiments. Statistically significant differences from % apoptotic cells in untreated cells (standard t-test; P < 0.05) are marked by “*.” D: HL60 cells were untreated or treated with 10 µM enzastaurin for 48 h. Where appropriate, cells were pre-treated with caspase-3 inhibitor (Z-DEVD-FMK) for 1 h before adding drug. Cell apoptosis was assessed by flow cytometry after Annexin V staining. Error bars represent the mean ± SD from three independent experiments. Statistically significant differences from % apoptotic cells in untreated cells (standard t-test; P < 0.05) are marked by “*.”
Enzastaurin-Induced Apoptosis is Independent of PKCβ in OCI-AML3 Cells
To determine if enzastaurin-induced cell death requires suppression of PKC β, OCI-AML3 cells were co-treated with another PKC β inhibitor (i.e., Calbiochem PKC β inhibitor). The IC50 values for PKC α, PKC βI, and PKC βII for the Calbiochem PKC β inhibitor are 331, 21, and 5 nM, respectively. We used Calbiochem PKC β inhibitor at 200 nM (to suppress both PKC β isoforms but not PKC α alone or in combination with 10 µM enzastaurin. The Calbiochem PKC β inhibitor alone had no effect on growth or survival of the OCI-AML3 cells even after 72 h (Fig. 3). As shown in Figure 3, enzastaurin suppressed cell growth of OCI-AML3 cells and inclusion of Calbiochem PKC β inhibitor had no effect on enzastaurin-induced cell killing.

Effect of PKC β inhibitor on enzastaurin-induced cell death in OCI-AML3 cells. OCI-AML3 cells were treated with vehicle (0.1% DMSO; marked 0), 10 µM enzastaurin, 200 nM calbiochem PKC β inhibitor, or a combination of 10 µM enzastaurin and 200 nM calbiochem PKC β inhibitor for 24, 48, or 72 h. Cell death was assessed by trypan blue exclusion assay.
Enzastaurin Blocks Phosphorylation of PKC α and ERK in AML Cell Lines
Enzastaurin was developed as an inhibitor of PKC β [Teicher et al., 2002a]. While the drug exhibits fairly high specificity for the kinase, enzastaurin is effective at inhibiting other PKC isoforms at higher concentrations. We examined expression and phosphorylation status of PKC α, PKC β, and PKC δ in the human AML-derived cell lines HL60 and OCI-AML3 (Fig. 4). Both HL60 and OCI-AML3 cells express abundant levels of PKC β. In both HL60 and OCI-AML3 cells, the kinase was potently phosphorylated suggesting high PKC β activity levels. However, enzastaurin up to 10 µM concentrations appeared to have little if any effect on PKC β phosphorylation levels in either cell line (Fig. 4). While enzastaurin displays high specificity for PKC β, the drug can inhibit PKC α at higher concentrations. The reported IC50 for PKC α is 0.039 µM. While little phosphorylation of PKC α is detected in OCI-AML3 cells, there is potent phosphorylation of the kinase in HL60 cells. As shown in Figure 4, enzastaurin potently blocked PKC α phosphorylation in both cell lines. At least in OCI-AML3 cells, the drug appears to suppress expression of the kinase at the highest dose used (i.e., 10 µM). These findings suggest that PKC α might be a possible target for the drug in AML cells.

Effect of enzastaurin on PKC α, PKC β, and PKC δ phosphorylation status in two AML cell lines. Phosphorylation status of PKC α, PKC β, and PKC δ was examined in HL60 and OCI-AML3 cells that were untreated or treated with varying doses of enzastaurin for 24 h. Western blot analysis was performed on protein lysate (1 × 106 cell equivalents) using antibody against p-PKCα, PKCα, p-PKCαβ, PKCβ, p-PKCδ, PKCδ, p-ERK, ERK, and actin.
Enzastaurin Blocks Membrane Localization of PKC α but not PKC β in AML Cell Lines
While phosphorylation of PKC is not truly indicative of its activation state, lipid activation of PKC results in its localization to cellular membranes [Gallegos and Newton, 2008; Newton, 2010]. We examined the effect of enzastaurin on PKC α and PKC β sub-cellular localization in OCI-AML3 cells using immunofluorescence (IF) microscopy. Cells were treated with 1 or 10 µM drug or vehicle (i.e., 0.1% DMSO) for 24 h. PKC α or PKC β was visualized using rabbit polyclonal antibody against each kinase and fluorescent labeled (i.e., Alexa 488) donkey-anti-rabbit antibody (PKC appears green in Fig. 5). DAPI was included to visualize nuclei (appears blue in Fig. 6). As shown in Figure 5, both PKC α and PKC β are distributed among intra-cellular membranes in vehicle-treated control cells. Enzastaurin at 1 µM suppressed localization of PKC α while the drug at even 10 µM was not effective at disrupting PKC β sub-cellular localization (Fig. 5). These results suggest that enzastaurin can inhibit activation of PKC α but not PKC β in OCI-AML3 cells.

Effect of enzastaurin on PKC α and PKC β membrane localization in OCI-AML3 cells. Membrane localization of PKC α and PKC β was examined in HL60 and OCI-AML3 cells that were treated with vehicle (0.1% DMSO), 1 µM, or 10 µM enzastaurin for 24 h. Cells were visualized using IF microscopy following staining with PKC α or PKC β antibody and Alexa 488 tagged secondary antibody (appears green). Cells were also stained with DAPI to visualize nuclei (appears blue).

Effect of enzastaurin on BCL2 phosphorylation in OCI-AML3 cells. A: OCI-AML3 cells were labeled with [32P] orthophosphoric acid (32P). BCL2 was immunoprecipitated using anti-sera from Santa Cruz Biotechonolgy, electrophoresed using 12% SDS/PAGE, transferred to an nitrocellulose filter, and phosphorylated bands detected by autoradiography. The identity of the BCL2 32P-labeled band was confirmed by Western analysis on the same filter using a different BCL2 antibody (from Dako). B: OCI-AML3 cells were treated with vehicle (0.1% DMSO; marked 0) or 10 µM enzastaurin for 24 h. Where appropriate, 20 nM okadaic acid was added to cells prior to addition of vehicle or enzastaurin. Western blot analysis was performed on protein lysate (1 × 106 cell equivalents) using antibody against p-BCL2 serine 70 and BCL2. C: OCI-AML3 cells were treated with vehicle (0.1% DMSO; marked 0) or 10 µM enzastaurin for 24 h. Where appropriate, 20 nM okadaic acid was added to cells prior to addition of vehicle or enzastaurin. Cell death was assessed by trypan blue exclusion assay.
Enzastaurin Blocks BCL2 Phosphorylation
It has been demonstrated in hematopoietic and acute leukemia cells that BCL2 phosphorylation at serine 70 is required for the anti-apoptotic molecule's full and potent suppression of apoptosis [May et al., 1994; Ruvolo et al., 1998; Deng et al., 2000]. Physiologic BCL2 kinases that promote survival include PKC α [Ruvolo et al., 1998] and ERK [Deng et al., 2000]. OCI-AML3 cells exhibit robust basal levels of phosphorylated BCL2 and phosphorylation of the anti-apoptotic molecule positively promotes resistance to chemotherapeutic drugs including the BH3 small molecule inhibitor ABT737 [Konopleva et al., 2002; Konopleva et al., 2006]. To determine the effect of enzastaurin on BCL2 phosphorylation, OCI-AML3 cells were treated with 10 µM enzastaurin for 3 h during metabolic labeling with 32P-orthophosphate and the phosphorylation status of BCL2 was examined following immunoprecipitation (Fig. 6A). Western analysis demonstrates that roughly equivalent levels of BCL2 protein were immunoprecipitated from untreated cells and cells treated with the drug. While BCL2 was highly phosphorylated in OCI-AML3 cells, phosphorylation of the protein was nearly completely inhibited by enzastaurin (Fig. 6A). Enzastaurin potently blocked both PKC α and ERK in OCI-AML3 cells (Fig. 4), thus it is not surprising that the drug is effective at blocking BCL2 phosphorylation. To determine if enzastaurin suppresses phosphorylation of BCL2 at serine 70 [the site targeted by PKC α and ERK; Ruvolo et al., 1998; Deng et al., 2000], we used a phospho-serine 70 BCL2-specific antibody to examine BCL2 phosphorylation by Western analysis in cell lysates of OCI-AML3 cells treated with enzastaurin (Fig. 6B). Cells were treated with 10 µM enzastaurin and/or 10 nM okadiac acid for 24 h. Protein phosphatase 2A (PP2A) dephosphorylates BCL2 at serine 70 and inhibition of the enzyme with okadaic acid promotes phosphorylation at this amino acid residues [Deng et al., 1998; Ruvolo et al., 1999]. Cells displayed basal levels of phosphorylated serine 70 BCL2 and levels were increased when treated with okadaic acid alone (Fig. 6B). The slower migrating band seen in the Western in cell treated with okadaic acid alone likely reflects a hyperphosphorylated form of BCL2 that results when other amino acid residues (e.g., serine 87 or threonine 69) are also phosphorylated [Deng et al., 2001]. As shown in Figure 6B, enzastaurin was found to suppress phosphorylation of BCL2 at serine 70 in OCI-AML3 cells and was effective at preventing the induced phosphorylation seen with okadaic acid. To determine if suppression of PP2A can protect cells from Enzasataurin, cell viability was assessed by trypan blue exclusion in OCI-AML3 cells treated with vehicle (0.1% DMSO) for 24 h, 10 nM okadaic acid for 24 h 30 min, 10 µM enzastaurin for 24 h, or pre-treated with 10 nM okadaic acid for 30 min prior to addition of 10 µM enzastaurin for 24 h. As shown in Figure 6C, okadaic acid did not protect OCI-AML3 cells from the drug.
Enzastaurin has Both Positive and Negative Effects on Protein Phosphorylation in HL60 Cells
While BCL2 is an important regulator of cell survival [Yang and Korsmeyer, 1996; Deng et al., 2001; Letai, 2008], it would be expected that enzastaurin's ability to suppress a number of PKC isoforms would result in suppression of phosphorylation of a broad number of proteins involved in survival signaling. To identify potential targets of enzastaurin in HL60 cells, two-dimensional electrophoresis and Western analysis of serine/threonine phosphorylation was performed on protein lysate from HL60 cells treated with vehicle (0.1% DMSO) or 10 µM enzastaurin for 24 h. Isoelectric focusing was carried out using 2% pH 3.5–10 ampholines (LKB/Pharmacia, Baltimore, MD) for 9,600 V/h in a tube gel which was then electrophoresed using a 10% acrylamide SDS slab gel. Duplicate gels were obtained from each sample and scanned with a laser densitometer and the images were analyzed. Student's t-test values were generated for an n of 4 gels to identify significant changes in spot density. Of >200 spots analyzed, 25 spots demonstrated a twofold decrease in phosphorylation with enzastaurin treatment (Table III). Conversely, 23 proteins showed a twofold increase in phosphorylation with enzastaurin treatment suggesting that non-PKC kinases may be activated in response to the drug (Table IV). An image of a representative gel is shown in Figure 7. Details of the protein's MW and pI and fold change in phosphorylation status with enzastaurin treatment are presented in Table III and Table IV.
| Spot number | pI | MW | Enzastaurin spot, % | Control (vehicle) spot, % | ENZA versus control difference |
|---|---|---|---|---|---|
| 3 | 6.2 | 175,185 | 0.098 | − | +++ |
| 77 | 7.6 | 63,866 | 0.043 | − | +++ |
| 94 | 7.6 | 59,125 | 0.046 | − | +++ |
| 121 | 8.4 | 55,543 | 0.129 | − | +++ |
| 257 | 7.8 | 30,361 | 0.762 | − | +++ |
| 260 | 7.9 | 29,324 | 0.638 | − | +++ |
| 299 | 7.6 | 21,843 | 4.462 | − | +++ |
| 1 | 6.1 | 205,059 | 0.476 | 0.086 | 5.5 |
| 68 | 8.6 | 65,137 | 1.722 | 0.469 | 3.7 |
| 96 | 6.3 | 59,088 | 0.120 | 0.046 | 2.6 |
| 102 | 6.4 | 58,697 | 0.100 | 0.032 | 3.1 |
| 107 | 6.4 | 57,525 | 0.047 | 0.015 | 3.0 |
| 108 | 6.1 | 57,329 | 0.125 | 0.034 | 3.7 |
| 109 | 6.3 | 57,264 | 0.037 | 0.012 | 3.1 |
| 118 | 6.1 | 55,832 | 0.124 | 0.043 | 2.9 |
| 134 | 6.1 | 53,943 | 0.245 | 0.060 | 4.1 |
| 224 | 5.4 | 38,963 | 0.122 | 0.044 | 2.8 |
| 233 | 6.8 | 36,685 | 0.038 | 0.015 | 2.6 |
| 251 | 7.7 | 31,721 | 0.980 | 0.344 | 2.8 |
| 266 | 5.7 | 28,733 | 0.235 | 0.074 | 3.2 |
| 275 | 6.8 | 28,028 | 0.080 | 0.030 | 2.6 |
| 291 | 6.9 | 25,836 | 0.077 | 0.028 | 2.7 |
| 300 | 7.7 | 21,604 | 2.582 | 0.839 | 3.1 |
- The vehicle used as control is 0.1% DMSO.
| Spot # | pI | MW | Enzastaurin spot, % | Control (vehicle) spot, % | ENZA versus control difference |
|---|---|---|---|---|---|
| 316 | 7.4 | 186,875 | — | 0.366 | — |
| 317 | 6.9 | 105,693 | — | 0.089 | — |
| 318 | 8.8 | 42,597 | — | 2.603 | — |
| 319 | 8.6 | 22,596 | — | 0.576 | — |
| 320 | 8.0 | 20,976 | — | 0.181 | — |
| 321 | 8.5 | 20,857 | — | 0.028 | — |
| 322 | 5.1 | 18,264 | — | 0.209 | — |
| 2 | 7.4 | 192,070 | 0.017 | 0.223 | −13.4 |
| 16 | 6.1 | 108,291 | 0.204 | 0.589 | −2.9 |
| 49 | 5.7 | 74,297 | 0.042 | 0.323 | −7.6 |
| 50 | 5.8 | 73,653 | 0.171 | 0.468 | −2.7 |
| 51 | 5.6 | 73,524 | 0.196 | 0.860 | −4.4 |
| 86 | 8.1 | 61,496 | 0.024 | 0.122 | −5.1 |
| 88 | 7.5 | 60,590 | 0.032 | 0.094 | −3.0 |
| 148 | 5.2 | 51,478 | 0.032 | 0.145 | −4.5 |
| 153 | 5.2 | 49,971 | 0.004 | 0.043 | −9.7 |
| 180 | 7.4 | 46,363 | 0.007 | 0.021 | −3.2 |
| 183 | 5.5 | 46,171 | 0.004 | 0.012 | −2.8 |
| 199 | 7.6 | 42,994 | 0.017 | 0.058 | −3.5 |
| 205 | 7.5 | 41,957 | 0.011 | 0.035 | −3.3 |
| 216 | 8.4 | 40,450 | 0.605 | 2.146 | −3.5 |
| 276 | 8.2 | 27,764 | 0.145 | 0.430 | −3.0 |
| 284 | 7.3 | 26,749 | 0.026 | 0.084 | −3.2 |
| 308 | 6.1 | 18,376 | 0.017 | 0.045 | −2.7 |
| 324 | 8.6 | 15,617 | 0.038 | 0.485 | −12.9 |
- The vehicle used as control is 0.1% DMSO.
- pI, isoelectric point; MW, molecular weight; ENZA, enzastaurin.

Representative 2D gel depicting the effect of enzastaurin on protein phosphorylation in HL60 cells. Polypeptide spots present only in enzastaurin (ENZA)-treated cells increased in ENZA-treated cells by 2.6-fold or greater are outlined in black. Protein molecular weight markers are shown on the right. The pH values determined at gel tube ends following isoelectric focusing are shown at the bottom. See Tables III and IV for spot identifications and measurements.
Enzastaurin Promotes Apoptosis in Primary Blast Cells Derived from AML patients
We next examined the ability for enzastaurin to effectively kill primary blast cells derived from AML patients. In vitro treatment of blast cells derived from AML patients demonstrated that enzastaurin potently killed cells. Treatment of AML blast cells with 5 µM enzastaurin for 48 h induced apoptosis in sensitive cells as indicated by an increase in Annexin V staining compared to vehicle-treated cells (Fig. 8A). As shown in Figure 8B, of nine AML blast cell treated with enzastaurin, five were sensitive to the agent. PKC α and PKC β protein expression and phosphorylation status in the blast cells from the nine AML patients were examined by Western analysis (Fig. 8C). Patients 1, 2, 4, and 7 exhibited robust phosphorylation of PKC α and/or PKC β. Interestingly, three of these patients (i.e., patients 1, 4, and 7) showed the least sensitivity to enzastaurin as indicted by Annexin V staining (< 5%; Fig. 8A). While the significance of expression of these PKC isoforms and sensitivity to the drug is not clear, the data does suggest that the drug can be effective at killing blast cells in a subset of AML patients and sensitivity might be influenced by PKC activity.

Effect of enzastaurin on cell viability and apoptosis in blast cells derived from nine AML pateints. Blast cells derived from nine (patients 1–9) patients were untreated or treated with 10 µM enzastaurin for 48 h. Cell apoptosis was assessed by flow cytometry after Annexin V staining (A). Percent surviving cells (B) was assessed by determining the ratio of viable treated cells to viable untreated cells. C: Phosphorylation status of PKC α and PKC β was examined in blast cells from nine AML patients. Western blot analysis was performed on protein lysate (1 × 106 cell equivalents) using antibody against p-PKCαβ, PKCα, PKC β, and tubulin.
DISCUSSION
The cPKC family members PKC α and PKC β play important roles in promoting cell survival and chemoresistance in leukemia cells [Redig and Platanias, 2007, 2008]. PKC β has been shown to be important in the development of colon cancer [Sauma et al., 1996; Murray et al., 1999; Zhang et al., 2004] and plays key roles in B-cell development. The kinase is important in B-cell receptor and Notch signaling and thus likely plays a role in lymphoid cancers [Redig and Platanias, 2007]. It was logical that enzastaurin as a PKC β inhibitor would be used as an anti-neoplastic agent for B-cell lymphomas and it is not surprising that the drug has shown promise in treating this disease [Robertson et al., 2007; Ma and Rosen, 2007]. While there is a well-founded rationale for using enzastaurin in treating lymphoid malignancies, the question arises whether targeting PKC β in myeloid diseases would be similarly effective. Gene expression of PKC β is significantly elevated in a number of leukemia cell lines (Fig. 1A) and in blast cells from newly diagnosed AML patients compared to counterpart cells (i.e., CD34+) from normal bone marrow donors (Fig. 1B). Enzastaurin promoted apoptosis in two AML-derived cell lines (Fig. 2A–C) and primary AML blast cells (Fig. 8A) but only at concentrations well above the IC50 for the suppression of PKC β. Consistent with the notion that the drug acted on a target other than PKC β, a specific PKC β inhibitor from Calbiochem showed no toxicity to OCI-AML3 cells and the drug did not affect toxicity of enzastaurin (Fig. 3). A recent article by Meng et al. [2010] suggest that inhibition of PKC β by means including the use of enzastaurin supports death-ligand induced apoptosis in AML and other leukemia cells. Furthermore, that study found that phorbol ester-mediated protective effect against death-ligand induced apoptosis in leukemia cells appears to require PKC β [Meng et al., 2010]. Perhaps PKC β plays a greater role in modulating the extrinsic apoptotic pathway in AML cells. Considering that enzastaurin is effective at inhibiting other PKC isoforms at concentrations used in clinical trials [Graff et al., 2005], the drug may be effective at suppressing PKC kinases that are relevant for AML cell survival such as PKC α [Kornblau et al., 2006]. PKC α suppresses apoptosis induced by HDAC inhibitors in a variety of multidrug-resistant leukemia cells [Castro-Galache et al., 2007]. IL-3 supports PKC α activation and promotes cell survival by mechanisms involving a number of pro-survival molecules such as AKT and BCL2 [Farrar et al., 1985; May et al., 1994; Blalock et al., 1999; Li et al., 1999; Deng et al., 2001; Redig and Platanias, 2007]. PKC α is emerging as an important prognostic factor in AML, particularly in the context of BCL2 and other pro-survival kinases [Kornblau et al., 2006; Kurinna et al., 2006]. The cPKC inhibitor Go6976 has been found to be effective at killing AML cells though it cannot be ruled out that inhibition of non-PKC kinases (e.g., JAK2) may be involved [Grandage et al., 2006]. Enzastaurin had little effect on PKC β phosphorylation (Fig. 4) or PKC β localization (Fig. 5). The drug, however, was effective at suppressing PKC α phosphorylation (Fig. 4) and PKC α membrane localization (Fig. 5), and the drug prevented phosphorylation of the PKC α substrate BCL2 (Fig. 6A,B).
The clinical trials with enzastaurin have been promising [Herbst et al., 2007; Robertson et al., 2007; Ma and Rosen, 2007; Oh et al., 2008; Morschhauser et al., 2008; Kreisl et al., 2009; Hanauske et al., 2009]. In the various studies, the drug has been well tolerated with few severe side effects reported. Among the 55 patients in the diffuse large B cell lymphoma study conducted by Robertson et al. [2007], there were no deaths reported and only one patient suffered hypomagnesemia (the sole grade 4 toxicity observed). Importantly, the drug was effective at delaying disease progression in a number of the patients and three patients exhibited complete responses [Robertson et al., 2007]. PKC activity in general has been associated with resistance to therapy in AML [Kornblau et al., 2006] so targeting PKC would be useful for AML therapy. Enzastaurin at high doses may be most effective in combination with other AML therapeutic drugs as a means to suppress PKC α and suppress BCL2 anti-apoptotic activity. Enzastaurin has been used successfully in combination with paclitaxel and other chemotherapeutic agents in mouse xenograft models [Teicher et al., 2002b; Ma and Rosen, 2007].
Enzastaurin was found to suppress phosphorylation of a distinct set of proteins (Table III). These proteins likely represent targets of PKC α or β or a kinase that is regulated by PKC. An example of a pro-survival kinase that is suppressed by enzastaurin inhibition of PKC is AKT [Graff et al., 2005]. In addition, PKC α has been found to negatively regulate the PP2A isoform (i.e., one containing the PPP2R5A subunit) that serves as the BCL2 phosphatase and promotes apoptosis in acute leukemia cells (Jiffar et al., 2004). It is possible that enzastaurin promotes activation of this PP2A isoform to suppress protein phosphorylation. The finding that the drug was effective at suppressing BCL2 phosphorylation (Fig. 6A,B) supports this notion. It was also found that enzastaurin promoted phosphorylation of a distinct set of proteins (Table IV). If enzastaurin supports activation of a specific PP2A isoform, then it is plausible that other PP2A isoforms will be suppressed since there is a competition between PP2A B subunits to associate with the enzyme's catalytic core [Silverstein et al., 2002; Strack et al., 2004; Ruvolo et al., 2008]. Perhaps the proteins displaying increased phosphorylation are the substrates of PP2A isoforms that compete with the PP2A subunit containing PPP2R5A. However, it is premature to implicate PP2A in this phenomenon as one cannot rule out the effect of the drug on non-PP2A protein phosphatases.
In summary, enzastaurin was found to be effective at killing AML-derived cells from cell lines and from primary blasts though by a mechanism that likely does not involve the inhibition of PKC β. The drug suppressed PKC α phosphorylation and localization and blocked activation of ERK and phosphorylation of BCL2. It is possible that enzastaurin promotes activation of a protein phosphatase that would be negatively regulated by PKC. These results suggest that enzastaurin could have clinical anti-leukemia activity in AML.
Acknowledgements
The authors would like to thank Eli Lilly and Company for providing enzastaurin and research support for this work.

