

Chemical Process Development in the Pharmaceutical Industry in Europe—Insights and Perspectives from Industry Scientists
Graphical Abstract
What are the tasks of chemical process development in the pharmaceutical industry? What are trends and challenges? This Scientific Perspective written by members of the European pharmaceutical industry introduces key concepts of drug development and key drivers for the future of chemical process development.

Abstract
Chemical process development is a critical component in the development process for active pharmaceutical ingredients (APIs). With interfaces to drug discovery and API manufacturing, chemical process development activities must deliver scalable, safe, cost-efficient, sustainable, and reliable processes for novel as well as marketed APIs. Despite its importance for the pharmaceutical industry and society, the domain of chemical process development, together with its advances and challenges, is often not well known to nonpractitioners. As industry scientists, we provide a scientific perspective on the state of affairs in chemical process development, which we believe will be of value to a broader audience, including academic researchers, students, and professionals from related fields.
1 Introduction
In 2024, plant and process chemists from leading European pharma companies celebrated their 25th anniversary. Founded as “Erfahrungsaustausch–Arbeitskreis Wirkstoffe” (Exchange Meeting for Active Pharmaceutical Ingredient Manufacturing), the first exchange took place in 1999 at Boehringer Ingelheim, one of the main drivers of the initiative. From this first event on, up to nine companies met annually with scientific experts from process chemistry, analytics, and quality assurance. All discussions are precompetitive in nature and focus on overarching trends and solution designs. The hot topics of small molecule (also referred to as new chemical entity, NCE) development and supply during preclinical phases up to market authorization have formed the agendas of these 2-day meetings. A recent example of a hot agenda topic is the challenge of identifying and controlling nitrosamines.[1] More general topics have been around technology trends, digitalization, new chemical modalities, and compliance with changes in the regulatory environment (regulatory authority expectations). As such, the “Erfahrungsaustausch” is providing a unique perspective on the status quo and trends in chemical process development.
2 Chemical Process Development in the Pharmaceutical Industry
Chemical process development activities in the pharmaceutical industry are centered around developing candidate molecules and marketed drugs. The term chemical process development is not sharply defined and typically covers multiple aspects such as chemical route scouting, scale-up, process optimization, solid-state determination, analytical method development, and quality control. It is therefore obvious that chemical development spans a broad range of domains and areas of expertise. In this perspective article, we will focus on aspects of drug substance development, which covers the unformulated active pharmaceutical ingredient (API). Equally important is drug product development which centers around the optimal formulation and delivery method for the API. The drug substance/drug product interface is of key importance in chemical development since several characteristics of the drug substance, like, for example, solid-state properties, impact drug product development.
In pharmaceutical companies, chemical process development functions are operating at a key interface between drug discovery (research) and API manufacturing (production). The main goal of any chemical development function is to enable fast, sustainable, safe, and cost-efficient access to chemical modalities, whether in clinical development or already approved as drugs (life cycle management activities). More specifically and depending on the development state of a given project, the goals can be, for example, to a) provide fast scale-up and supply of candidate molecules for preclinical studies or b) to develop improved processes for high-volume projects in order to improve sustainability and cost efficiency or c) to engage in transfer activities of chemical manufacturing processes into internal or external manufacturing plants (Figure 1).

We purposefully use the term chemical modalities in this article to account for a significant broadening of development pipelines in the pharmaceutical industry. This broadening beyond classical small molecules includes synthetic modalities such as oligonucleotides, peptides, or drug conjugates, to name a few. Most pharmaceutical companies today follow a “modality-agnostic” approach in drug discovery, meaning that the choice of modality is driven by the pharmacological target and not vice versa. This requires chemical process development organizations to constantly evaluate whether their skill and technology foundation is fit for purpose to support a highly diverse and dynamic development pipeline.
Another key driver for organizations to stay up to date with regard to expertise and technology is the steadily increasing molecular complexity of small molecule development candidates.[2, 3] A recent pipeline analysis at Sanofi, which we consider exemplary for the pharmaceutical industry, is depicted in Figure 2. Molecular complexity, which is defined by multiple parameters such as number of stereogenic centers, has increased considerably, calling for state-of-the-art synthesis and manufacturing capabilities.

Over the last decade, most pharmaceutical companies have also significantly increased their collaboration network with external contract development and manufacturing organizations (CDMOs), which provide chemical intermediates or even end-to-end process development and manufacturing services. This has become possible due to a rapidly growing number of global service providers that can offer services with a high degree of quality (for example, good manufacturing practice (GMP) qualified materials),[4] speed, and cost efficiency. Given this external opportunity landscape and the dynamic evolution of pharmaceutical pipelines, chemical process development functions need to carefully balance internal and external investments (“make or buy”).
Chemical process development organizations are an integral part and key value driver in the pharmaceutical industry. Through chemical and technological innovation, they turn complex drug candidate molecules into viable business cases and supply clinical trials under high time pressure.
Also, for approved drugs on the market, stable, and high-quality supply is of utmost importance to earn and keep the trust of patients and authorities.
3 Accelerated Approvals—Speeding Up Drug Development
The global innovation landscape within the pharmaceutical industry has significantly changed over the past decades. In addition to the large integrated pharmaceutical companies as traditional players, many small and mid-size biotechnology companies have entered the stage. Backed by venture funding and leveraging a global CDMO landscape, these biotechnology companies can bring their development candidates to clinical trials or even through market authorization, often with a focus on rapid clinical proof of concept.[5, 6] At the same time, expedited clinical development programs have become feasible in several therapeutic areas with high medical need, such as oncology. For example, the US Food & Drug Administration (FDA) Accelerated Approval program allows earlier approval of innovative therapies for serious conditions based on clinical surrogate parameters. Such approaches can significantly shorten development timelines and reduce complexity of clinical development programs, which is of great importance, especially for small and mid-size biotechnology companies.
As a result, we observe an ever-increasing competition around therapeutic areas and pharmacological targets. The importance of speed and time to market was recently analyzed: Comparing 104 products launched since 2010 and analyzing value captured based on launch order as well as therapeutic advantage, it became obvious that first to launch products capture larger market share and most value, even if they do not show the highest therapeutic advantage.[7] The implications for chemical development organizations are manifold: In the past, they have focused on ensuring rapid drug substance delivery for preclinical studies as well as clinical phase 1 supply. This was often done via a “fit for purpose” approach whereby focus was on scalability of the synthetic route (often the available route from the discovery stage) and optimization of the final synthesis steps (typically most critical with regard to quality control).[8, 9] Within standard development times, further changes to the synthetic route, including change of starting materials, could be done during later clinical development phases. Within accelerated approval frameworks, development timelines are significantly shortened so that a more holistic understanding of the synthetic route and planning of process R&D activities with the end (regulatory submission) in mind is critical.[10] Emphasis must be placed on engaging early in lead optimization campaigns, often long before a development candidate is advanced to preclinical development. Goals of this early engagement are to familiarize oneself with the available synthesis routes and identify starting materials as well as critical improvements with a special focus on the final synthetic steps. Also, understanding the solid-state landscape of the API and selecting a suitable crystal form and potentially salt form can have dramatic benefits for the whole clinical development program. The authors have seen impressive examples of such fast development projects, but it must be stated that there are also significant risks associated with frontloading of activities. Development organizations need to carefully balance their resources across the portfolio. Significant frontloading at risk almost inevitably comes at the expense of slowing down other portfolio projects. The years to come will show to what extent regulators will raise the bar for expedited projects demanding more proof of therapeutic advantage. For the time being, the authors consider such fast development projects rather the exception than the norm of clinical development pipelines.
Streamlined clinical development programs with smaller patient populations often reduce the amount of drug substance needed for clinical development. At the same time, acceptable treatment cost in small patient populations with high medical need can be higher. Therefore, the initial importance of cost of goods and process scalability is often lower compared to speed and further process improvements to drive down cost may be performed after launch.
A highly instructive example for accelerated development and its challenges is the KRASG12C inhibitor divarasib (1) from Roche/Genentech. Divarasib (1) is currently in late-stage clinical development and forms part of a larger group of innovative KRASG12C inhibitors such as Sotorasib (Amgen), Adagrasib (Mirati), and BI-0474 (Boehringer Ingelheim). Structurally, divarasib (1) poses considerable synthetic challenges such as a rotationally hindered heterobiaryl axis (atropoisomers), a reactive acrylamide warhead as covalent binding unit, and a highly functionalized pyridine motif. The retrosynthetic analysis is depicted in Scheme 1.

Divarasib (1) is derived from a late-stage insertion of the acrylamide moiety as well as a crucial atroposelective Negishi coupling between quinazoline 2 and aminopyridine 3.[11] These key disconnections were already featured in the first-generation synthesis of divarasib (1). The most selective ligand identified for the first-generation process was a research compound that was not available on a larger scale at that time. The better available Walphos ligands showed acceptable conversions but decent atroposelectivities. A chiral chromatographic purification was required to achieve the desired purity of 9 (Scheme 2). While the process was now fit for purpose to supply initial kilogram quantities, further scale-up to multi-100 kg quantities was deemed not to be feasible, mainly due to the need for chromatography. As part of the second-generation process development, the following process adaptations were made: 1) A continuous flow process for the efficient synthesis of quinazoline 2 at scale,[12] 2) a significantly improved access to aminopyrimidine 3 based on readily available starting materials,[13] 3) an operationally simple and easily scalable access to precursor 8 featuring low process mass intensity (PMI),[14] and 4) an improved catalyst with (R,R)-chiraphite as ligand for the Negishi coupling avoiding the hitherto necessary chromatographic purification.[15] An overview of the second-generation process can be seen in Scheme 2.

4 CDMO Landscape and Supplier Steering
As mentioned above, both the complexity of APIs and their synthesis lengths have increased significantly over the last decades, driving pharmaceutical companies to outsource part of their activities in chemical development. On the one hand, this is purely driven by the additional, fluctuating capacity needed for labs and manufacturing plants, and on the other hand, by the need to access highly specialized technologies not available (yet) internally.
A synthesis typically starts from purchasing chemicals readily available from supplier catalogues. In contrast, outsourcing syntheses of API-specific building blocks to external CDMOs requires know-how transfer and close cooperation, including appropriate legal frameworks to safeguard intellectual property (IP) and to ensure high quality and safety standards are applied. Typical contracts are Master Service Agreements (MSA) and Quality Assurance Agreements (QAA). Furthermore, chemical development is rather a journey than a straightforward road. Ongoing activities need to be closely monitored and guided cross-functionally by internal experts (for example, chemists, analytical experts, quality assurance, engineers). This does not only include regular (virtual) updates but also technical visits, complemented by quality (GMP) and safety (EHS) audits. This is especially crucial when the process is to be transferred later on, either to the process-owning company (sponsor) or to another CDMO capable of supplying late-stage clinical phases or even the market. Hence, building and growing expert teams overseeing externalized activities while maintaining strong collaboration with internal teams is key.
Companies pursue individual outsourcing strategies, defining the scope of outsourcing during the product lifecycle. The supplier landscape for nonregulated materials (Figure 3, blue window) is generally more diverse than for GMP steps.

In fact, most big pharmaceutical companies have a short list of preferred GMP suppliers with a good collaboration track record.
The choice of the partner depends on different factors, for example, capabilities, capacities, technologies, and location as well as own assets and the package to be outsourced: is only material to be ordered, or the process development itself? How cost-sensitive is the current asset? Is the technology required standard, or is it only available at a limited number of specialized CDMOs? Does the company have a long-term strategic partner? Can and should the whole CMC development be carried out at one partner? At what (clinical) phase is the project currently?
At larger pharmaceutical companies, the final (GMP) chemical steps toward the API were initially kept in-house, while more basic chemistry starting from commodities was outsourced. Smaller pharmaceutical and biotech companies typically have a higher outsourcing ratio due to the lack of internal capacity. As globalization accelerated in the 2000s, a huge landscape of suppliers emerged worldwide, especially in eastern Asia. India and, especially, China have developed a competitive industry characterized by highly qualified personnel and huge capacities. In the meantime, not only catalogue chemicals but most custom-made intermediates are manufactured in Asia at quality levels comparable to originator companies. Very recently, increasing geopolitical strains have been leading to a re-evaluation of outsourcing strategies, including re-balancing footprint across regions.
5 Regulatory Framework for API Development and Manufacturing
The pharmaceutical industry is among the most regulated businesses. When developing and manufacturing an API for clinical trials or market supplies, there are several regulatory requirements that must be met to ensure the safety, quality, and efficacy of a drug. These regulatory requirements are outlined in numerous guidelines and regulations.
The major quality framework is named Good Manufacturing Practice (GMP), which is essential for the manufacturing of APIs for human use.[4] The associated guidelines broadly describe the quality management in various areas like personnel training, buildings and facilities, process equipment, and documentation.
Furthermore, a manufacturing process must undergo a validation stage prior to routine manufacturing, as pharmaceutical companies must demonstrate that their processes are capable of consistently producing products that meet predetermined specifications and quality attributes. This also includes a continuous process verification during the whole lifecycle of a drug.
Along these lines, a process chemist is typically closely connected with an analytical chemist in the development and scale-up stage to jointly build an analytical control strategy combined with an in-depth process understanding in preparation for process validation and regulatory filing. Among others, guiding questions in this stage are: what is the origin, fate, and purge of impurities that could have a potentially negative effect on patients? What are the proven acceptable ranges and the critical process parameters that have influence on the quality of the final API? What are the critical quality attributes for the starting materials? What is needed to control the physicochemical parameters of an API and what is the stability profile of the API under various conditions?
Participating companies of the “Erfahrungsaustausch” each follow systematic approaches (Quality by Design, QbD) to build holistic control strategies and compile the requisite documentation for new drug approvals. Meetings of the “Erfahrungsaustausch” are intended to exchange best practices, new regulatory trends, and challenges in implementing the latter. To this end, deep understanding of the science is a key success factor to lead these discussions and guide risk-based decisions. Documentation of a new drug application is highly regulated and typically referred to as chemistry, manufacturing, and controls (CMC) dossier.
The regulatory frame of a drug development journey defines the content of the work packages and the timing of activities calculated backwards from the launch date (Figure 1).
Along these lines, key operational parameters for chemical processes and all analytical methods needed for characterization are ideally frozen for a standard API project at least 1–2 years prior to the launch date. In this final development stage, an in-depth process understanding is created, a validation campaign is conducted, and long-term stability data are collected with material from the final process.
There is a constant trend to accelerate drug development programs with the goal to provide life-saving cures to patients much faster (Section 3). Accordingly, current standard development timelines of ≥10 years are under scrutiny, and this puts pressure on an earlier chemical process freeze and leaves less room to identify the best chemical route with respect to costs and process efficiency. At the same time, process robustness, safety, and quality must not be at risk at any time.
Moreover, the regulatory pathways for new modalities such as peptides or antibody–drug conjugates (ADCs) are different from those of traditional small-molecule-based drugs. Companies must anticipate more authority interactions and longer timelines for approvals, as the dossiers need to be amended to reflect the different nature of the new modality drugs.
All of these regulatory guardrails clearly define the playing field for a process chemist to translate science into patient solutions.
6 Sustainability: From Green Chemistry to Eco Footprint Reduction
Reducing the resource consumption and waste generation of chemical processes, as well as avoiding hazardous chemicals, have always been intrinsic goals of chemical process development in the chemical and pharmaceutical industry, as it goes hand in hand with significant production cost reduction and increased process safety. The topic of green chemistry was boosted by the postulation of concepts like atom economy[16] and the 12 principles of green chemistry[17] some decades ago. Today, sustainability has become a “license to operate” topic for the whole industry, including pharma, and additional effort is needed that goes beyond cost reduction. Medicines and innovative treatments have a tremendous positive impact on patients, but their manufacturing still contributes to global warming and climate change.
All major pharmaceutical companies have defined ESG (environment, social, and governance) targets, including significant reductions of resource consumption and CO2 emissions. These targets will be hard to achieve without the consideration of production processes for APIs. As these goals can only be reached using the best science, the pharmaceutical industry together with chemical societies (for example, the ACS Green Chemistry Institute), founded consortia like the ACS GCI Pharmaceutical Roundtable (ACS GCIPR) in 2005. This collaboration has generated a multitude of contributions to the topic of sustainable chemistry and engineering, including guides and tools.[18] One important outcome is easy-to-use solvent guides[19-22] by consortia and individual companies, which are widely applied in development laboratories as early as in preclinical R&D. Guidance is also given by the European Union's REACH Substance of Very High Concern (SVHC) or by the discussion of Polyfluorinated Alkyl Substances (PFAS). A simple sustainability KPI that has been adopted early throughout the industry and is routinely applied is the process mass intensity (PMI),[23] which is the total mass of materials employed per mass of product. Usually less than 1% of materials used during production are incorporated into an API and PMIs can typically range from 100 kg kg−1 of API to multi-thousand kg/kg, depending on the length of the synthesis and the starting point of the calculation. By allocating the contribution of reagents, organic solvents, and process water to their specific share of the PMI, it allows targeted discussion and comparison of processes. A recent example of the great improvement of the PMI that is possible by chemical process R&D is the development of a multi-kilogram scale route for a chiral spirocyclic isoxazolone (Scheme 3).[24] Here, a complex 6-step racemic synthesis for a building block for multiple projects in the field of oncology was replaced by a much shorter, enantioselective route, resulting in an impressive 98% reduction of the PMI measures from over 5000 kg of waste to close to 100 kg.

The total product carbon footprint of a chemical process is contributing to CO2 emissions and global warming. Therefore, more accurate and comprehensive life cycle assessments (LCA) are required. LCAs are a complex task due to a lack of clearly defined standards and availability of data. More sophisticated methodologies have been proposed to assess chemical syntheses (for example, PMI-LCA and iGal 2.0), which take environmental indicators into account that go beyond simple PMI assessments.[25-27] This may include efficiency of synthetic processes, more detailed categorization of raw material (fossil, renewable, precious metals) and waste (for example, inorganic and organic), chemical hazards, and carbon footprint. The consumption of material is closely linked to the carbon footprint, and for pharmaceuticals, it is usually dominated by the bill of purchased materials (Figure 4).[28, 29] Accurate data for carbon footprints of many raw materials, commodities, or intermediates are often not available. Decarbonization of the whole supply chain is a major lever to decrease product carbon footprints.

As depicted in Figure 5, the bill of materials of pharmaceutical processes is dominated by solvents.[30, 31] The overall contribution of organic solvents to the carbon footprint not only results from their production but is also connected to energy consumption during an API production process like heating, cooling, and distilling reaction mixtures where solvents are the main component. In addition, CO2 is released during incineration of solvent waste for energy recovery and large quantities of solvent are needed for the purpose of equipment cleaning.[32, 33] Traditionally, reuse and recycling of solvents played a minor role in pharmaceutical production—not least due to the comprehensive quality regulations and the risk of the accumulation of process-specific impurities. Nevertheless, many companies are now considering systematic solvent recycling and the associated increased effort for analytical evaluation and risk assessment, for example, concerning nitrosamines. Reduction of use, recovery, reuse, and recycling of solvents must become a prime focus to reduce product carbon footprints of APIs.

The “green solvent” water also needs to be carefully assessed, as ecotoxic contaminations may rule out more environmentally benign biological wastewater treatment and determine energy-consuming “incineration” of aqueous waste.[34, 35]
Short, convergent, and efficient chemical routes, process intensification, and application of highly selective transformations all add tremendously to sustainability—putting new scientific advances in fields like chemo-, biocatalysis, electrochemistry, and photochemistry into the spotlight. Unmet scientific needs have recently been highlighted by academic and industrial members of the SusChem Switzerland platform to trigger research with a high sustainability impact.[36]
The discussion around more sustainable production is particularly intense in the field of new modalities such as antibody–drug conjugates, synthetic peptides, and oligonucleotides. Currently, these classes of drugs show metrics (PMI) up to ≫20 times higher than those of classic small molecules, though relating it to dose, treatment cycle, and overall benefit for the patient may place this number in a better perspective.[37]
In summary, there is still a need for standardized methods for sustainability assessments in the industry and comprehensive data availability for purchased materials. High project attrition rates in combination with shortened time-to-market timelines are an additional challenge for effectively steering resources in order to achieve comprehensive sustainability improvements in specific R&D projects. Sustainability is today a process attribute demanded by boards and associations for modern medicinal products, and it is being pursued with great urgency. Driving sustainability and green chemistry process development plays an important role for achieving the net zero climate goals of pharmaceutical companies in the upcoming years.
7 New Chemical Modalities
Despite the success of “classical therapies” predominantly based on small molecules (SMOLs) and therapeutic antibodies, there is still a high unmet medical need in key therapeutic areas. Therefore, discovery scientists are constantly expanding the pharmacological target space, thanks to a boost in available methods to interrogate the human genome and proteome. Molecules that often serve these new modes of action and go beyond classical small molecules are typically referred to as “new modalities” by discovery scientists.[38, 39] The major classes that are typically discussed in this context are proximity-induced degraders, oligonucleotides, peptides, radiotherapeutics as well as bioconjugate formats such as ADCs.
We conducted a survey among member companies of the “Erfahrungsaustausch” on the relevance of new modalities in their specific development portfolios. As concluded from Figure 6, ADCs have been named as the leading category with the highest relevance, followed by peptides and oligonucleotides (“TIDES”), while radiotherapeutics and degraders are focus topics for a subgroup of member companies only.

Which challenges are associated with these new drug formats from the viewpoint of a process chemist? Foremost, the molecular structure of the new modalities defines the selection of technologies needed for synthesis and manufacturing (Table 1). For example, a solid-phase synthesis platform to manufacture peptides and oligonucleotides is needed at all intended scales, as this is still the major methodology to assemble such molecules. For purification and isolation, one cannot solely rely on classical crystallization procedures as used in common downstream protocols for small molecules (SMOLs), but additional access to different preparative chromatographic methods is needed, and isolation is typically performed via spray and freeze drying.
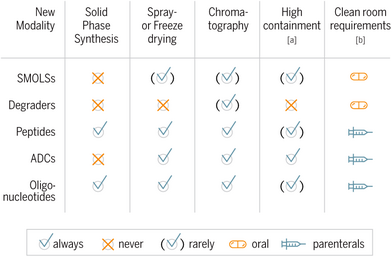 |
- a) High containment infrastructure, which is needed to handle highly potent active ingredients (HPAI).
- b) Complexity of clean room class infrastructure mainly depends on the route of pharmaceutical administration.
Furthermore, the majority of new modalities are intended for parenteral applications mainly due to their physicochemical properties. As a direct consequence, the manufacturing of the active ingredient intended for human use has to take place in an advanced clean room class environment as defined by the regulators, such as EMA or FDA. These regulations aim at a control of bioburden and avoidance of contamination of the active ingredient, for example, by foreign particles. This translates into a need for a sophisticated clean room infrastructure, for example, with orchestrated air locks and ventilation systems and monitoring protocols deeply embedded into the GMP quality systems. As we intend to protect our patients from harm, the same holds true for our operators in the manufacturing suits. In particular, the assembly of highly potent molecules on a technical scale requires organizational and technical measures to protect the operator and to comply with health and safety regulations. For instance, antibody drug conjugates are typically classified as highly potent and key manufacturing steps are therefore operated in closed systems such as glove box environments (see Figure 7).

Table 1 shows an overview of typical unit operations, technologies', and infrastructure requirements needed to drive a new modality portfolio in a process chemistry setting. Between the new modality classes, you find synergies and overlapping themes in the requirements for unit operations and infrastructure, which offers options for a multiple use of platform elements. In the end, companies have to make choices to guide their investments into internal capabilities and build-up of scientific excellence or engage with external partners and specialized custom manufacturers.
Overall, the arrival of new modalities has added a new layer of molecular diversity to the drug development arena with direct impact on the technical requirements and skills in the development stages. In the following paragraphs we discuss the challenges and opportunities in more detail, and we focus on peptides, oligonucleotides, and ADCs as these are the modalities of most strategic relevance for member companies of the “Erfahrungsaustausch.”
Following pioneering work such as the industrialization of 36mer HIV entry inhibitor Fuzeon in 2003, synthetic peptides have been revived in recent years as an important therapeutic modality for the treatment of numerous diseases. Their unique properties as medium-sized molecules to address challenging drug targets have significantly increased the interest in novel peptide therapeutics[40] and peptide conjugates[41] in multiple therapeutic areas during the last decade. Peptides account for approximately 5% of the global pharmaceutical market, with global sales higher than US$50 billion already in 2019.[42] A recent push in large-scale manufacturing of therapeutic peptides can be attributed to the advent of GLP-1 agonists, mimicking intestinal hormones for effective treatments of Type 2 diabetes and/or obesity. Such peptides usually include linear chains of 30–40 amino acids, decorated with fatty acid side chains and noncanonical amino acids to modify their pharmacological half-life. These molecules qualify for both synthetical or recombinant or even hybrid manufacturing methods (see below), and next to several already market-approved examples, Figure 8 shows the Phase III development compound survodutide (11) from Boehringer Ingelheim.[43]

This recent trend toward an increasing number of peptide therapeutics has a significant impact on chemical development units as the development of routes for commercial large-scale manufacturing of peptides comes with specific challenges that drive the exploration of novel technologies for synthesis and purification.
Many recently designed synthetic peptides contain noncanonical amino acids[44] in order to optimize their pharmacokinetic and binding properties as well as their cell permeability.[45] As a consequence, chemical development units face the task of rapidly coming up with novel approaches toward these important building blocks in a cost-efficient and sustainable way. Among others, biocatalytic processes hold great promise as they often proceed with high stereocontrol and without the need for protecting group manipulations.[46] Recently new biocatalytic transformations have been described that provide access to β-branched noncanonical amino acids[47] and to valuable α-tertiary amino acids.[48]
Solid-phase peptide synthesis (SPPS) is routinely used to produce peptides in the early discovery phase during optimization of hits for potency and stability toward lead candidates. In early drug discovery, automated synthesizers are routinely used to generate peptide libraries at milligram scales.[49] When programs further progress toward preclinical and clinical studies, larger amounts (>10 g) of APIs are often needed. These first supplies are often prepared by manual batch operations as it allows higher flexibility and better control in comparison with automated procedures. Recently a continuous-flow (CF) SPPS workflow to optimize and deliver multigram quantities of peptide fragments has been described.[50]
In general, large-scale chemical supply of peptide therapeutics is commonly achieved using two methods: solid-phase peptide synthesis (SPPS) and liquid phase peptide synthesis (LPPS). SPPS generates a linear sequence, while LPPS allows for linear and convergent syntheses. A hybrid approach combining SPPS and LPPS has been used for the kilogram-scale manufacture of GLP-1 agonist tirzepatide, where short fragments were synthesized via SPPS and then connected together using LPPS.[51] Recombinant biotechnology and enzymatic assembly can be efficient alternative approaches to chemical peptide synthesis but have limitations, for example, regarding noncanonical amino acids.[52]
When selecting a synthesis strategy, factors such as material demand, batch size, manufacturing capability, complexity, scalability, automation potential, starting material availability, costs, and regulatory control strategy need to be considered. Using LPPS for manufacturing shorter peptides (5–10 amino acids) offers advantages such as the utilization of different protecting groups and the opportunity for process optimization to limit material and reagent usage and to reduce impurity formation.[53] However, LPPS requires more manual interactions during production and may involve higher initial costs and longer process development timelines. SPPS technology is widely used and reliable, with established building blocks, supply chains, and known chemistry conditions.
SPPS often makes use of excess solvents and reagents, which negatively impact the environment. In a recent perspective article, the manufacturing processes for peptide APIs have been comprehensively assessed, and the impact of synthesis, purification, and isolation stages on the Process Mass Intensity (PMI) metrics was discussed. According to this analysis solid-phase peptide synthesis (PMI ≈ 13 000) does not compare favorably with other modalities such as small molecules (PMI median 168 − 308) and biopharmaceuticals (PMI ≈ 8300).[54] This holds true despite cases as discussed in Section 6 where low patient doses lower the waste generated per patient. According to this analysis, liquid-phase peptide synthesis tends to have more favorable average PMI values compared to solid-phase peptide synthesis. These high PMI values clearly demonstrate the urgent challenge for chemical development units to develop greener processes[55] and to come up with novel technologies for synthesis and purification in peptide chemistry.[56] Given the rapidly growing development portfolio and the higher API amounts needed, more sustainable[57] and efficient ways for the commercial manufacturing of peptide therapeutics will be a key success factor.
Over the past several decades, the use of synthetic oligonucleotides as a therapeutic modality gained tremendous momentum, and consequently, nearly 20 nucleic acid therapeutics have been approved for treatment of various diseases to date. Their modes of action include antisense oligonucleotides (ASOs), splice-switching oligonucleotides (SSOs), and small interfering oligonucleotides (siRNAs). All of these oligonucleotides share a need for improved pharmacological properties compared to unmodified RNA and DNA. Major modifications of oligonucleotides brought to market thus far contain just a handful of first- and second-generation modifications, among them 2′-fluoro RNA, 2′-O-methyl RNA, 2′-O-(2-methoxyethyl) RNA (MOE), and the phosphorothioates. These multi-1000 Da APIs are manufactured by a sophisticated, semi-automated, and high-yielding solid-phase synthesis, followed by cleavage from the solid support, chromatographic purification, and finally isolation by, for example, freeze drying. In the case of siRNA, an additional annealing step is required. While the first assets were more in niche applications needing only small quantities of API, common diseases like cardiovascular indications requiring manufacturing of tons of API per year are more and more in focus nowadays. However, this is a challenge with the current manufacturing technologies, which allow only relatively small batch sizes of several kilograms. An example is the recently approved drug Leqvio, which is shown in Figure 9: Inclisiran (Leqvio, 12) is a GalNAc-conjugated siRNA.[58, 59] The focus of process development is mainly on how to establish a scientifically sound manufacturing and analytical control strategy that is in line with current regulations for small molecules. Dedicated teams with experts in oligo synthesis, automation, and large-scale purification, as well as mass spectrometry (MS) experts mastering the sophisticated analytical MS/MS techniques, are needed to successfully register these unconventional products.

Among the most successful new modality formats are ADCs with 14 registered drugs, showcasing that this concept has grown from a scientific curiosity to an established therapeutic concept.[60, 61]
The common molecular design principles for bioconjugates are based on a targeting moiety combined via a linker with a functional small molecule-based “warhead.” In this context, an antibody or peptide format serves as a homing unit, and it is designed to have a high specificity for the targeting tissue. The warhead typically consists of a cytotoxic small molecule.
The role of a chemical development function is focused on manufacturing of the warhead, the linker, and the conjugation process to assemble the components to the final drug substance (the ADC itself). Along these lines, a couple of challenges are associated with these activities.[62]
The manufacturing of ADCs comprises a multitude of production steps. Important elements are, for example, the fermentation and downstream processing of an antibody, the multistep synthesis of a warhead and a linker, the conjugation to form the drug substance and final sterile filling to provide the drug product for clinical applications. Along the value chain, several in-process-control and release tests, freeze-thaw steps, and shipping between manufacturing sites have to be orchestrated. Accordingly, the lead time for a clinical batch easily sums up to periods of 12–18 months. This is a particular challenge for an organization in a development environment with speed and flexibility as important value drivers.
In general, ADCs are classified as biologics from a regulatory standpoint as an antibody format is incorporated in the structure. Biologics must be characterized more extensively during process development and for marketing approval to account for their structural complexity. This is even more relevant in the case of random conjugation chemistry, which yields statistic mixtures of conjugates. Accordingly, the width of the analytical method toolbox is quite elaborate, spanning different chromatographic and mass spectrometry methods up to functional cell-based assays to prove consistent quality and potency of the batches produced. In most cases, ADCs are highly active molecules and the payloads in their pure forms can even have occupational exposure limits (OEL) far below 1 µg m−3. Accordingly, organizational and technical measures such as glove-box technologies (see Figure 7) need to be in place to ensure an adequate containment level for the final manufacturing steps.
An illustrative example is Roche/Genentech's ADC polatuzumab vedotin (Polivy, 13), which was approved in 2019 in the US for the treatment of diffuse large B-cell lymphoma in adults (Figure 10). It is prepared by conjugation of the payload-linker vcMMAE to the monoclonal antibody anti-CD79b. The warhead (auristatine, MMAE) is manufactured in 20 chemical steps from readily available raw materials. The last steps of the payload synthesis are conducted under GMP due to the high activity of MMAE in a high containment facility. The linker is a protease-cleavable valine-citrulline (VC) dipeptide maleimide linker and is prepared in seven chemical steps and subsequently coupled to the payload via amide bond formation. Conjugation of the payload-linker to the monoclonal antibody proceeds via thiol-maleimide Michael addition and is typically done in a plant for biologics manufacturing.

New modalities are complementing today´s classical small molecule portfolio in member companies of the “Erfahrungsaustausch.” This pipeline enrichment comes with challenges', especially for a technical development unit.
In general, there is clear regulatory guidance for conventional small molecules, while there are less regulations for next-generation therapeutics lacking established procedures and processes during a registration process. This is a challenge as insufficient CMC documentation poses a large risk during the regulatory approval process. Additionally, scale-up and industrialization of the new drug formats are often uncharted territory in chemistry, analytics, and engineering.
As a mitigation, companies need to build critical mass in resourcing, competencies, and adequate infrastructure complementing the classical small molecule landscape—be it internally or with partners.
8 Novel Technologies
Drug discovery and development have constantly been enabled by advances in synthetic organic chemistry as well as novel technologies associated with drug substance manufacturing.
Examples include biocatalysis and transition metal catalysis that today belong to the standard capabilities of every major chemical development organization and have changed the way that process chemists devise synthetic routes to target molecules.
Biocatalysis utilizes biological macromolecules such as enzymes to catalyze chemical transformations.[63] Improving existing enzymes by enzyme engineering has enabled the development of modified nonnatural enzymes that show better efficiency, selectivity, or stability compared to the natural (wild-type) enzymes or even catalyze novel transformations and cascades of enzymatic transformations.[64] For exploring enzyme activity and for creating new and efficient biocatalysts, the combination of directed evolution and rational enzyme design using computational tools and high-throughput experimentation is applied. Several leading pharmaceutical companies have built up internal enzyme engineering capabilities and workflows, while others rely on CROs or institutes to evolve their enzymes. In an illustrative example from Novartis, a highly engineered phenylalanine ammonia lyase (PAL) was developed, which was then used to implement a more efficient alternative route to a key intermediate of Olodanrigan (14, Scheme 4).[65] Even though no conversion of cinnamic acid substrate could be detected in initial screening experiments with wild-type PAL enzymes, presumably because of the bulky and electron-rich substituents on the substrate, an extensive enzyme engineering program was initiated for the directed evolution of an enzyme starting from an engineered variant from Anabaena variabilis specifically for the conversion of cinnamic acids. The new route applying the engineered enzyme was found to be superior in terms of cost and sustainability to the current route, which used asymmetric Rh-catalyzed hydrogenation. The engineered enzyme variants were obtained after several cycles of enzyme evolution and more than 20 amino acid mutations from the starting point.

Transition metal catalysts can activate substrates and accelerate reactions through various mechanisms, including coordination, ligand substitution, insertion, and elimination. In combination with high-throughput screening and applying machine learning, a broad range of chemical transformations can be run metal-catalyzed, aiming for shorter chemical routes and greener processes. In the last two decades, carbon– and carbon–heteroatom cross-coupling reactions such as Suzuki-, Buchwald-Hartwig-, Negishi-couplings and others emerged as two of the most important transformations for process chemists, also having a significant impact on the design of drug substance synthesis. While in the former century, poly-heterocyclic motifs were typically built up one by one using classical heterocyclic and condensation chemistry, this is nowadays conveniently done by transition metal-catalyzed cross-coupling. An illustrative example is the synthesis of Roche's development candidate Fenebrutinib (15), as can be seen in Scheme 5. The eight-ring core is built up from two three-ring starting materials in three Pd-catalyzed cross couplings.[66]

Given the more recent significant developments in areas such as photoredox catalysis, C–H activation, and electrochemistry, one might wonder whether these methods are already routinely used in chemical process development. Currently there are only a few examples of their implementation on a larger scale (>100 g). This is in stark contrast to the area of drug discovery, where especially C–H activation and photoredox catalysis are more commonly used. What are the reasons for this discrepancy?
As pertinently discussed by colleagues from Merck, several hurdles need to be overcome when scaling up these methods.[67] In the case of photoredox catalysis, careful reactor design and selection of light source are among the most common challenges. An instructive example is immersion well (IW) reactors that have been developed at Bayer (Figure 11). These reactors enable the rapid scale-up of heterogeneous reaction mixtures that are often difficult to handle in flow reactors.[68]

More generally speaking, however, the design of reactors as well as the prediction of reaction parameters is mostly done case by case and is often limited to molecules of medium complexity. Reactor designs with broad applicability and predictability of reaction parameters will further facilitate the adoption of photoredox catalysis in chemical process development.
Electrochemistry, which makes use of electrons as versatile reagent, holds the promise of efficient and sustainable transformations, especially if green energy sources are employed. Given the recent surge in electrochemical method development, the question about applications in chemical process development arises.[69, 70] Again, careful reactor design that considers electrode and membrane material, electrode geometry, current density, pressure, temperature, and solvent is of critical importance for successful scale-up.[71]
A representative example is the synthesis of Finerenone (16), a mineralocorticoid receptor antagonist developed by Bayer.[72] The synthetic route toward Finerenone (16) is depicted in Scheme 6.

In order to render the synthesis more efficient and sustainable, recycling of the undesired enantiomer was envisioned. Among other options, an oxidation/reduction sequence making use of an electrochemical reduction step was investigated. The realization of this step on production scale was a multiyear endeavor that ultimately led to the design of a pilot electrochemistry flow reactor depicted in Figure 12.

As can be seen from these two examples, efficient and broadly applicable scale-up procedures in the field of photoredox catalysis and electrochemistry are still lacking. This makes the integration of such methods into development or even market supply campaigns a time-consuming and expensive endeavor, which can become prohibitive if viable alternative synthetic routes exist. Nevertheless, we are optimistic that with further innovations in reactor design and more published examples of large-scale applications associated with better data-driven predictions of reaction parameters on scale, the adoption of these novel technologies in chemical process development will significantly increase.
9 Automation and Digitalization
Upon exploring current literature or engaging in interdisciplinary discussions, one might imagine a future dominated by autonomous chemical process development, powered by artificial intelligence (AI) predictions and robotic execution. Despite significant strides in AI and automation, such a scenario remains largely elusive, even in the distant future.
Technology advancements in the arena of process chemistry can range from new chemical methodologies, over new reactors to better analytical methods. The digital value chain can be organized in three parts: (a) data ingestion of lab devices, reactors, plants, and machines; (b) data storage and handling according to FAIR (“Findable, Accessible, Interoperable, and Re-usable”) data principles; and (c) modelling, prediction, and simulation of experiments and reactors (for example, digital twins). Hence the key question is, in which areas of process chemistry do we see the biggest opportunities to disrupt the way we are developing drug candidates?
First, by leveraging fast in silico-based experiments to thereby decrease time-intensive wet lab work: As the ability to predict the outcome of scientific experiments purely in silico is considered the ultimate goal for AI applications in chemical development, this field has seen significant focus and progress in recent years. Given that these methods require large volumes of data for training, the attention has been on specific unit operations of chemical processes (for example, yield prediction) or molecular properties (solubility, pKa, polymorph landscape) where substantial data is available.
Second, if wet lab experiments need to be performed, planning of experiments can be assisted by machine learning methods like Bayesian optimization (or when it comes to design space characterization, conventional design of experiments (DoE) approaches)[73] to allow for selection of most rewarding experiments. Wet lab execution can be augmented by sophisticated lab automation such as robot arms, sampling robots, solids and liquid handling, and workflow apps. Ultimately, a combination of these approaches allows for semi-autonomous chemical reaction screening and optimization campaigns that can be run overnight and on weekends.
Third, as far as an individual wet lab experiment is concerned, maximizing the value of each experiment has become more important than ever, meaning to record and pull all relevant data out of an experiment by applying sophisticated devices, probes, and apps. Often large language models or AI assistance are expected to find important hints and nonobvious correlations that are potentially missed by a human. However, in our experience, the key challenge here is to strike a balance between important process insights and flooding project teams with too much useless data. How might a future workflow look like, given the focus areas mentioned?
With regard to planning relevant wet lab experiments, the user-friendly Bayesian optimization framework BoFire is noteworthy. This framework was co-developed in an open collaboration between Evonik, BASF, Boehringer Ingelheim, and Imperial College. It enables implementation of Bayesian optimization within our organizations, effectively eliminating one-factor-at-a-time-based optimization methodologies (Figure 13).[74, 75]

To maximize the value of each experiment, the chemistry team at Bayer, in collaboration with external partners, developed the MOCCA framework.[76] This framework automates HPLC-DAD (diode array detection) raw data analysis. Its key features include peak deconvolution and peak tracking for starting materials, products, and side components. This aids in better understanding large reaction screening datasets while freeing our scientists from repetitive tasks (Figure 14).

These concepts and workflows are regularly applied in our development projects, and we believe this is just the beginning of what is possible. However, we also see limitations with regard to scaling these applications within our organizations:
First, machine learning (ML) methods require large, publicly often unavailable, datasets to create meaningful models. Consequently, academic groups develop these methods using smaller (and often imbalanced) datasets, leading to overly optimistic results. When applied to our larger internal datasets, these methods often underperform due to the increased complexity of the chemical space. Various potential solutions exist for this challenge. These include enhancing high throughput experimentation (HTE) capabilities in academia, fostering closer collaborations between academia and industry, and encouraging a cultural shift in the pharmaceutical industry to share more data publicly. Important data initiatives like the Open Reaction Database are crucial steps toward making more consistent data publicly available. However, although larger datasets will lead to more realistic assessments of model performance and potentially more extrapolative models, further development of new models, especially hybrid ones taking expert knowledge into account, is of great importance.
Second, model implementation is challenging due to nonstandardized usage of complex libraries and lack of focus on usability, including documentation and user interface elements. Future developments like the “ChemProp” framework for molecular modeling and open-source frameworks like “BoFire” will simplify model implementation.
Third, significant process steps are not yet widely covered by ML models. Process chemists spend 70% of their time optimizing workup and purification, yet AI support for these steps is limited. Recent industry advancements in miniaturizing and standardizing work-up optimization screenings may be a solution, particularly for generating relevant training data.[77]
10 People, Skills, and Organizational Setup
As we alluded to in previous sections, process chemistry departments today face a broad set of challenges. These range from increasing quality and regulatory requirements over more pressure regarding cost of goods for APIs (manufacturing cost) to elevated project management challenges, quite often triggered by accelerated development timelines that include multiple project plans and require various supply scenarios. Also, innovation both in terms of chemical methods as well as technology continues at high speed.[78] One example is new chemical modalities that require special skills with regard to synthesis and purification. At the same time, most process development departments within the pharmaceutical industry have seen significant reductions in internal workforce and a higher reliance on external CDMOs. What are the implications regarding employee skills? First and foremost, it cannot be overstated that synthetic organic chemistry remains the core skill for process chemistry: A thorough mechanistic understanding of chemical processes and a creative mindset to perform root cause analyses and potentially identify alternative synthetic routes today can still decide the fate of a project. At the same time, we would like to underline those skills in additional fields such as analytical science, solid-state science or project management are of equal importance, but a more detailed discussion around them would be beyond the scope of this perspective. We acknowledge that chemistry students at universities receive a thorough education in organic chemistry. However, process chemistry is rarely part of university curricula or is covered as part of chemical engineering curricula. Chemistry students are therefore often unaware of the challenges and opportunities of process chemistry. We believe that university-industry collaborations such as mentorship programs, guest lectures by experienced industry scientists as well as internships could help to close this gap. As we have discussed in Section 9, data science applications become more and more integrated into the daily routine of process chemists. While we do not propose that every process chemist has to become a seasoned data scientist, there needs to be a good level of curiosity and willingness to integrate new data science methods in workflows. We do see great examples from our participating companies that have developed cutting-edge data science applications through collaboration of process chemists with academia.[79] Gratifyingly, there are also more and more academic groups pioneering data science applications in organic chemistry.[80]
To provide a better overview of required skills in industrial process chemistry departments, we conducted a survey among participating companies of the “Erfahrungsaustausch.” The resulting word cloud can be seen in Figure 15.

As the highest ranking can be seen, organic chemistry knowledge that has been highlighted earlier in this section. In the Mentimeter poll, it does not pop up so obviously as it is mentioned with five different expressions like “knowledge in chemistry” and “strong organic chemistry.” Chemistry is followed by learning agility, meaning the ability to quickly adapt to a changing environment and acquire new skills as well as proficiency in organic chemistry.
Perhaps not unexpectedly, there are also several soft skills, such as collaboration and communication mentioned by several participants. This underlines the importance of continued training and learning across all levels (lab technicians, scientists, and senior managers). Ultimately, the future of every process chemistry department will depend on the ability to hire talents and rapidly adapt its skill base to the changing environment.
In recent years, participating companies in the “Erfahrungsaustausch” have also adapted their organizational setups to achieve higher resource flexibility. Workforce reductions in process chemistry departments often did not go hand in hand with reduced project portfolios. This has sometimes led to difficulties in rapidly allocating proper resources to projects. Solution designs vary from larger team sizes (vs. micro teams) to more sharply defined roles (scientific experts, lab technician experts) to entirely new operating models. The success of these organizational frameworks needs to be assessed in the future and is a constant topic for discussion at the Erfahrungsaustausch.
In summary, chemical process development is a rapidly evolving field, and we are convinced that there are and will be plenty of opportunities for students and apprentices. They will continue to shape chemical process development in the future for the benefit of patients and societies.
Acknowledgements
The authors would like to thank Julia Bagińska and Dr. Finn Burg for their help and valuable comments during the creation of figures and chemical drawings.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.

