

Enantio- and Regioconvergent Nickel-Catalyzed Etherification of Phenols by Allylation to Access Chiral C(sp3)−O Allyl Aryl Ethers
Graphical Abstract
A nickel-catalyzed etherification of phenols with regioisomeric mixtures of racemic silylated and germylated allylic chlorides enables the enantio- and regioconvergent formation of unsymmetrical 1,3-disubstituted chiral allylic aryl ethers as a single regioisomer (see Scheme). Key to success is the metalloid moiety governing the regioselectivity, and these highly functionalized allylic ethers can subsequently undergo chemoselective chemical modifications.

Abstract
An enantio- and regioconvergent allylation of phenols under nickel catalysis with an α-/γ-regioisomeric mixture of racemic silylated/germylated allylic chlorides is reported. The silyl/germyl group governs the regioselectivity, and the transformation affords enantiomerically enriched unsymmetrical 1,3-disubstituted allyl aryl ethers with excellent regiocontrol in good yields and excellent enantioselectivities. Notably, no nickel-mediated C−O bond activation is observed at room temperature. The synthetic value of these densely functionalized silicon-containing building blocks is demonstrated in a series of chemoselective transformations, including a [3,3]-sigmatropic rearrangement for the construction of an α-chiral silane.
Oxygenated stereogenic carbon atoms are ubiquitous in synthetic chemistry, and chiral allyl aryl ethers are important representatives of that class of compounds.1, 2 Their synthesis by SN2 or even SN1 displacement of chiral allylic electrophiles is burdened by regioselectivity issues and erosion of stereochemical information. Transition-metal-catalyzed processes to overcome these challenges were developed over the past two decades, the majority of which rely on allylic substitution starting with branched or linear allylic electrophiles involving terminal π-allyl intermediates of ruthenium,3 rhodium,4 palladium,5, 6 and iridium6a, 7, 8 with oxygen nucleophiles (Scheme 1, top). Alternatively, branched allylic ethers can be synthesized with high enantioselectivity through a unique palladium(II)-catalyzed SN2’ reaction of allylic trichloroacetimidates.9, 10 Furthermore, the utilization of acyclic symmetrical 1,3-disubstituted allylic electrophiles for etherification is well established with palladium being the prevailing catalyst (Scheme 1, middle).6, 11 Conversely, achieving control over the regio- and enantioselectivity as well as the alkene geometry for unsymmetrical 1,3-disubstituted allylic ethers remains a challenge and is rarely documented.12, 13

Previously reported asymmetric allylic substitution reactions accessing chiral allylic C(sp3)−O motifs. Alk=alkyl, Ar=aryl.
Our laboratory recently demonstrated that bulky silyl14 and germyl15 groups in allylic electrophiles can steer nickel-catalyzed C(sp3)−C(sp3) bond formation away from the silicon- or germanium-substituted carbon atom to afford a single regioisomer. On this basis, we envisioned using this strategy to realize a stereoconvergent transformation of unsymmetrical 1,3-disubstituted allylic electrophiles for the construction of chiral allyl aryl ethers (Scheme 1, bottom).
Of note, Takeda and co-workers reported a racemic protocol for the synthesis of allylic ethers and thioethers under nickel catalysis almost twenty years ago.16 This isolated example is not only seminal for being a nickel-catalyzed O-allylation but also for showing that there is no competing nickel-mediated C−O bond activation17, 18, 19, 20 at a reaction temperature of 50 °C. The development of catalysis with nickel as an earth-abundant metal is highly desirable,21 and we here report a highly enantioselective allylic substitution with phenol nucleophiles that proceeds with superb regiocontrol.
We began our study by reacting Me2PhSi-substituted allylic chloride rac-1 a with phenol in the presence of a base under nickel catalysis (Table 1). A solvent screen revealed at an early stage that sodium phenoxide or phenol/base would react directly with the allylic electrophile in polar aprotic solvents such as DMA or DMF. With no participation of the chiral nickel catalyst, no enantioinduction yet good control of the regioselectivity was seen in the formation of product 3 aa. The competing SN2 reaction was completely suppressed in toluene, and the nickel-catalyzed allylic displacement with chiral Box ligand L1 facilitated the formation of product 3 aa in low yield but with excellent enantioselectivity as a single regioisomer (entry 1). No further improvement was achieved with other chiral ligands (see the Supporting Information for the complete set of tested ligands). For example, PyBox L2 led to a substantially higher yield but lower enantioselectivity (entry 2), and the use of QuinOx L3 did not result in any product formation (entry 3). In turn, the base had a dramatic effect on the yield while maintaining the high level of enantioselection (entries 4–6). Cs2CO3 instead of K3PO4 did improve the yield but NaH brought about the desired effect, reaching 71 % yield with >99 % ee for 3 aa; an organic base such as DBU was inferior to any of the inorganic bases that were evaluated.
|
|||
Entry |
Deviation from the standard conditions |
Yield [%][b] |
ee [%][c] |
|---|---|---|---|
1 |
none |
23 |
98 |
2 |
L2 instead of L1 |
74 |
79 |
3 |
L3 instead of L1 |
trace |
n.d. |
4 |
Cs2CO3 instead of K3PO4 |
39 |
98 |
5[d] |
NaH instead of K3PO4 |
71 |
>99 |
6 |
DBU instead of K3PO4 |
11 |
98 |
7[d] |
THF instead of toluene |
10 |
98 |
8[d] |
2-Me-THF instead of toluene |
64 |
90 |
9[d] |
(Ph3P)4Ni instead of Ni(cod)2 |
n.r. |
n.d. |
10[d] |
NiCl2/Mn (3.0 equiv) instead of Ni(cod)2 |
n.r. |
n.d. |
11[d] |
50 °C instead of RT |
23 |
99 |
12[d] |
w/ 15-crown-5 (1.2 equiv) |
61 |
90 |
13[d] |
NaOPh (1.2 equiv) instead of PhOH/NaH |
30 |
98 |
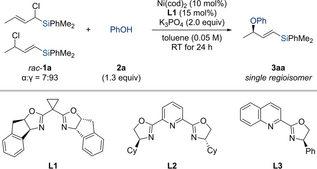
- [a] All reactions were performed on a 0.10 mmol scale. [b] Determined by GLC analysis with biphenyl as an internal standard. [c] Determined by HPLC analysis on a chiral stationary phase. [d] 1.3 equiv. of NaH used as the base and 12 h reaction time. n.d.=not determined. n.r.=no reaction. DBU=1,8-diazabicyclo[5.4.0]undec-7-ene. 15-crown-5=1,4,7,10,13-pentaoxacyclopentadecane.
We continued with the evaluation of other parameters (see the Supporting Information for the complete set of data). Even with NaH as the base, toluene was a better choice than THF and 2-Me-THF (entries 7 and 8). No reaction took place with (Ph3P)4Ni or NiCl2/Mn as the nickel precatalyst (entries 9 and 10). Elevating the reaction temperature was detrimental (entry 11). The addition of 15-crown-5 had no beneficial effect (entry 12). The high ee value was retained with preformed sodium phenoxide but the yield was poor (entry 13). The phenoxide is believed to be contaminated with traces of water, and a control experiment using three equivalents of water showed no product formation. This could hence explain why the efficiency is higher when the phenoxide is generated in situ.
As further optimization of the reaction setup in entry 5 was fruitless, we decided to proceed with the exploration of the scope (Scheme 2). The effect of the substitution pattern at the silicon atom was not particularly pronounced. Good yields and excellent enantioselectivities were obtained with trialkylsilyl groups such as Et3Si- as in 1 b, tBuMe2Si- as in 1 c, and BnMe2Si- as in 1 d; the allylic electrophile with the very bulky tBu2MeSi- group yielded trace amounts of product (not shown). Aryl groups at the silicon atom were compatible as seen from the model compound 1 a with Me2PhSi- and congener 1 e with MePh2Si-; the lower yield isolated for 1 f with the more sterically hindered Ph3Si- group as well as the markedly diminished ee value showed a limit of the methodology.

Scope I: Variation of the silyl and germyl group and the alkyl substituent. All reactions were performed on a 0.10 mmol scale. Isolated yields refer to analytically pure material after flash column chromatography on silica gel; yields in parentheses refer to the conversion. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. [a] A 51 % yield with 96 % ee was obtained on a 1.0 mmol scale.
A 1.0 mmol experiment was performed with the model substrate with slight decreases in yield and enantioselectivity (1 a→3aa). Elongation of the aliphatic R’ group in the electrophile was tolerated, furnishing the chiral allylic ethers in excellent enantioselectivities as a single regioisomers (1 g→3ga and 1 h→3ha). A phenyl group at the terminus of the linear alkyl chain was compatible depending on the distance to the reactive site; homobenzyl was fine (1 j→3ja) but benzyl was not with both yield and ee value marked reduced (1 i→3ia). We attribute this to steric hindrance, and this is in accordance with the experimental finding that the allylic displacement is thwarted with an isopropyl group in this position (not shown). Lastly, the protocol was also applicable to the corresponding germanium derivatives (4 a→5aa and 4 b→5ba).
Next, we investigated the substitution pattern of the phenol nucleophile in the model reaction (2 b–u; Scheme 3). The three regioisomeric cresols 2 b–d yielded the products 3 ab–ad with enantiomeric excesses of 90 % or higher; yield and enantioinduction were lowest for o-cresol (1 d) and highest for m-cresol (1 c). That result was validated by other phenol derivatives that bear two or one substituent in the ortho position (2 e→3ae and also 2 o→3ao and 2 u→3au); in the case of product 3 ae, we detected the α-regioisomer, again indicating that there are limitations for sterically congested reactants. However, other functional groups in the para-position such as tert-butyl (2 f), phenyl (2 g), methoxy (2 i), an acetal (2 k), a thioether (2 l), a fluoro group (2 m), and a boronate (2 n) delivered the allylic ethers in good yields and with high enantioselectivities. Furthermore, β-naphthol (2 h), 3,5-dimethoxyphenol (2 j), and a ketone-containing phenol (2 p) were fully compatible with the general procedure. Of note, electron-deficient phenol derivatives bearing a cyano or carboxyl group in the para-position did not react (not shown). A few simple naturally occurring phenols as well as a biologically active substance delivered the O-allylated products in good yields and excellent enantioselectivities (2 r–t→3ar–at and 2 q→3aq).

Scope II: Variation of the oxygen nucleophile. All reactions were performed with 0.10 mmol of the allylic chloride rac-1 a, 1.3 equiv. of the phenols 2 b–u, 10 mol % of Ni(cod)2, 15 mol % of L1, and 1.3 equiv. of NaH in toluene (2.0 mL) at room temperature for 12 hours. Isolated yields refer to analytically pure material after flash column chromatography on silica gel; yields in parentheses refer to the conversion. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. [a] r.r.=93 : 7. [b] d.r.=50 : 50. r.r.=regioisomeric ratio.
To demonstrate the pivotal role of the silyl group in steering the C(sp3)−O bond formation away from its proximity, we carried out two control experiments (Scheme 4A). First, rac-6 a with two linear alkyl chains afforded allylic ether 7 as a regioisomeric mixture with poor enantioselectivity. Replacing an alkyl chain with an aryl group, we conducted the reaction with cinnamyl chloride rac-8 a which once again furnished the product with poor regioselectivity and enantioselectivity. In light of nickel's ability to engage in C−O bond activation,17, 18, 19, 20 we performed a crossover experiment to examine the reversibility of the C(sp3)−O bond formation. When product 3 aa was subjected to the standard protocol in the presence of p-methoxyphenol (2 i), none of the crossover product 3 ai was detected; the allylic ether 3 aa was recovered without loss of the stereochemical integrity (Scheme 4B). This result rules out any reversibility of C(sp3)−O bond formation. A radical-trapping experiment with the addition of the radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) to the standard reaction conditions resulted in no product formation and the detection of the corresponding TEMPO adduct by HRMS (Scheme 4C). Note: A substrate for a radical-clock ring-opening experiment could not be isolated due to its chemical instability. Based on these control experiments, we propose the formation of an allyl radical that also explains the stereoconvergence (see the Supporting Information for the proposed catalytic cycle and a stereochemical model).22

Mechanistic studies. All reactions were performed on a 0.10 mmol scale. Isolated yields refer to analytically pure material after flash column chromatography on silica gel. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. PMP=p-methoxyphenyl.
The allylic ethers 3 are densely functionalized with several options for further chemoselective manipulations (Scheme 5). Cleavage of the aryl ether in 3 is the same as deprotection of an allylic alcohol. One-electron oxidative removal of the PMP group in 3 ai furnished the known allylic alcohol 10 in 86 % yield without any racemization. By comparison with reported data,23 the absolute configuration was assigned as R (see the Supporting Information for details). Additional transformations were done with allylic ethers 3 aa. Utilizing the vinylsilane unit in 3 as a linchpin, both a Hiyama cross-coupling to give 11 and a halodesilylation to yield 12 proceeded in moderate yields. When ICl was used instead of NIS, we found its addition across the C−C double bond rather than the Si−I exchange. Highly functionalized product 13 was obtained in good yield and with no loss in stereochemical integrity. We also achieved the racemization-free hydrogenation of the vinylsilane 3 aa to the alkylsilane 14 in 62 % yield. Subsequent oxidative degradation of the C(sp3)−Si bond in 14 following Fleming's procedure24 provided access to the reported alcohol 15 in 95 % yield. The expected absolute configuration of product 15 is in accordance with the literature.25 Lastly, transforming the chiral allylic aryl ether 3 aa into the α-chiral silane 16 by a Lewis acid-catalyzed Claisen rearrangement process enabled us to achieve the 1,3-chirality transfer and the desired C−C bond formation with slight erosion in enantioselectivity; the double bond geometry could not be controlled.11b

Product derivatization. Isolated yields refer to analytically pure material after flash column chromatography on silica gel. Enantiomeric excesses were determined by HPLC analysis on chiral stationary phases. [a] The relative stereochemistry of this acyclic molecule could not be assigned. [b] CAN (2.0 equiv), MeCN/H2O (3 : 1, 0.10 M), RT for 12 h. [c] (Ph3P)2PdCl2 (5.0 mol %), Ph3P (10 mol %), CuI (1.0 equiv), TBAF (3.0 equiv), PhI (1.2 equiv), DMF (0.10 M), 100 °C for 15 h. [d] NIS (3.0 equiv), MeCN (0.10 M), 60 °C for 15 h. [e] ICl (1.0 equiv), CH2Cl2 (0.10 M), 0 °C to RT for 1 h. [f] Pd/C (2.0 mol %), H2 (1 atm), MeOH (0.10 M), RT for 12 h. [g] Hg(OAc)2 (1.5 equiv), CH3CO3H (35 % wt in AcOH, 1.0 mL), RT for 12 h. CAN=ceric ammonium nitrate, NIS=N-iodosuccinimide, TBAF=tetra-n-butylammonium fluoride.
In summary, we disclosed an efficient protocol for the enantio- and regioconvergent etherification of regioisomeric mixtures of silylated and germylated allylic chlorides with phenol nucleophiles under nickel catalysis. The metalloid moiety is again the key to success as it governs the regioselectivity to furnish the unsymmetrical 1,3-disubstituted chiral allylic ethers as a single regioisomer. Furthermore, general methods for achieving C−O bonds under nickel catalysis have remained elusive so far, perhaps also because nickel is known to activate such bonds.17, 18, 19, 20 The synthetic value of the products was demonstrated by the cleavage of the PMP-protecting group to liberate the corresponding allylic alcohol and by other chemoselective transformations.
Acknowledgments
This research was supported by the Deutsche Forschungsgemeinschaft (Oe 249/25-1) and the State of Berlin (Elsa-Neumann-Scholarship for doctoral researchers to D.N., 2023–2025). M.O. is indebted to the Einstein Foundation Berlin for an endowed professorship. Open Access funding enabled and organized by Projekt DEAL.
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

