

Transition Metal-Free Catalytic C−H Zincation and Alumination
Graphical Abstract
A transition metal free catalytic C−H zincation and C−H alumination of heteroarenes using (β-diketiminate)-metal complexes, where the only by-products are H2 or CH4, is reported. This is achieved by coupling endergonic C−H metalation with subsequent exergonic dehydrocoupling, with the latter process proceeding by metal-specific pathways.

Abstract
C−H metalation is the most efficient method to prepare aryl–zinc and –aluminium complexes that are ubiquitous nucleophiles. Virtually all C−H metalation routes to form Al/Zn organometallics require stoichiometric, strong Brønsted bases with no base-catalyzed reactions reported. Herein we present a catalytic in amine/ammonium salt (Et3N/[(Et3N)H]+) C−H metalation process to form aryl-zinc and aryl-aluminium complexes. Key to this approach is coupling an endergonic C−H metalation step with a sufficiently exergonic dehydrocoupling step between the ammonium salt by-product of C−H metalation ([(Et3N)H]+) and a Zn−H or Al−Me containing complex. This step, forming H2/MeH, makes the overall cycle exergonic while generating more of the reactive metal electrophile. Mechanistic studies supported by DFT calculations revealed metal-specific dehydrocoupling pathways, with the divergent reactivity due to the different metal valency (which impacts the accessibility of amine-free cationic metal complexes) and steric environment. Notably, dehydrocoupling in the zinc system proceeds through a ligand-mediated pathway involving protonation of the β-diketiminate Cγ position. Given this process is applicable to two disparate metals (Zn and Al), other main group metals and ligand sets are expected to be amenable to this transition metal-free, catalytic C−H metalation.
Introduction
Catalyzed C−H functionalization is ubiquitous as it is a highly efficient way to increase molecular complexity.1 In this field, arene C−H borylation is particularly powerful,2 while C−H silylation is also a useful synthetic tool.3 In comparison, catalytic arene C−H functionalization processes that install more electropositive main group elements are extremely under-developed.4
Zinc- and aluminium organometallics are more nucleophilic than organo-boranes/-silanes and are widely used in synthesis,5 with aryl–zinc reagents ubiquitous due to the Negishi cross-coupling reaction.6 Methods that convert C−H bonds directly into C−Zn and C−Al bonds remain dominated by the use of at least one equivalent of a strong Brønsted base combined with a zinc/aluminium electrophile (Figure 1a).7, 8 In contrast to stoichiometric methods, catalytic C−H zincation/alumination is rare despite the higher efficiency of this approach. To our knowledge the only reports on catalytic arene C−H zincation and C−H alumination use a (β-diketiminate)ZnH9 and a (β-diketiminate)AlH2,10 respectively, with H2 formed as the only by-product (Figure 1b). Significantly, palladium catalysis was crucial in both these processes as <5 % C−H metalation was observed in the absence of the Pd catalyst. Notably, transition metal-free catalytic C−H zincation/alumination has not been reported to our knowledge, and would offer an attractive, orthogonal preparation compared to the established methods.

(a) C−H zincation/alumination using stoichiometric base. (b) Catalytic C−H zincation/alumination using a Pd-catalyst. (c) Our approach, C−H metalation using sub-stoichiometric R3N/[(R3N)H]+.
In developing new Al/Zn catalytic C−H metalation routes, it is instructive to consider the more established area of transition metal-free C−H borylation. In these reactions frustrated Lewis pairs (FLPs) are often used to effect C−H functionalization.11 During this process an electrophilic borane forms the C−B bond while a basic component (e.g., R3N) is protonated. The resultant Brønsted acid (e.g., [(R3N)H]+) then can react with a hydroborane forming H2 (termed dehydrocoupling herein). This second step can enable the process to be catalytic as it can regenerate the reactive boron electrophile and the base. Generally, with borane/base FLPs both the C−H borylation and dehydrocoupling steps are exergonic.12 Unlike the boron systems, analogous chemistry using Zn/Al electrophiles must overcome the challenge of weaker (vs C−B) C−Zn and C−Al bonds, which results in arene C−H metalation being endergonic. When incorporated into a catalytic cycle this endergonic first step will result in: (i) increased overall barriers for the catalysis once the second step (dehydrocoupling) is taken into account and (ii) a catalytic cycle that is less thermodynamically favored (and potentially endergonic). Therefore, we sought to develop Zn/Al FLPs13 that combined an endergonic FLP-mediated (hetero)arene C−H metalation step with a significantly exergonic dehydrocoupling step (e.g. between [(amine)H]+ and [Zn]−H/[Al]−H) to generate an overall exergonic process. With systems containing low barrier dehydrocoupling steps this would enable catalytic (sub-stoichiometric in amine/ammonium salt) C−H metalation as more of the key electrophilic metal complex will be formed post dehydrocoupling (Figure 1c).
Herein we report that a sub-stoichiometric quantity of Et3N/[(Et3N)H]+ enables the catalytic C−H zincation/alumination of heteroarenes using (β-diketiminate)metal hydrides/alkyls (Figure 1c, bottom) with H2/MeH the only by-product. Notably, the dehydrocoupling step in the C−H zincation process is only feasible due to ligand non-innocence. Combined, this work introduces a transition metal-free, catalytic route to form C−Zn and C−Al bonds directly from the heteroarene.
Results and Discussion
C−H Zincation Studies
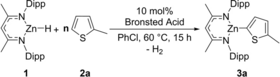
As part of our recent C−H borylation work we demonstrated that the reaction of NacNacZnH (NacNac={(2,6-iPr2C6H3)N(CH3)C}2CH) with an anilinium cation, [(DMT)H]+ (DMT=N,N-dimethyl-4-toluidine), to form H2 and [NacNacZn(DMT)]+ is exergonic.13d Initial computational studies indicated that DMT can be displaced from zinc by a heteroarene. The resultant Zn-(heteroaryl-H) complex and free DMT were then calculated to effect C−H zincation with feasible barriers (for solution reactivity). While this FLP-type C−H zincation was calculated to be endergonic, coupling it with a dehydrocoupling step (between NacNacZnH (1) and [(DMT)H]+) was explored as a route to make the C−H zincation process exergonic overall. Initially, NacNacZnH 1 and 2-methyl-thiophene 2 a, were combined with 10 mol % [(DMT)H][B(C6F5)4]. This led to 41 % conversion to the C−H zincation product 3 a after heating at 60 °C for 15 h in chlorobenzene (PhCl, Table 1, entry 1). Under identical conditions in the absence of the ammonium salt no C−H metalation was observed (entry 2).
|
|||
Entry |
Brønsted Acid |
Eq. 2 a |
Yield[b] (%) |
|---|---|---|---|
1 |
[(DMT)H][B(C6F5)4] |
1 |
41 |
2 |
– |
1 |
0 |
3 |
[(Et3N)H][B(ArCF3)4] |
1 |
68 |
4 |
[(Et3N)H][B(C6F5)4] |
1 |
60 |
5 |
[(Et3N)H][OTf] |
1 |
0 |
6 |
[(DMT)H][OTf] |
1 |
0 |
7 |
[(Et3N)H][B(ArCF3)4] |
2 |
78 |
8 |
[(Et3N)H][B(ArCF3)4] |
3 |
87 |
9[c] |
[(Et3N)H][B(ArCF3)4] |
2 |
88 |
- [a] 1 (0.1 mmol, 1.0 equiv.), 2 a (n equiv.), and Brønsted acid (0.01 mmol, 0.1 equiv) in PhCl (0.6 mL). [b] Yield by 1H NMR spectroscopy versus CH2Br2 added as internal standard at the end. [c] Reaction carried out for 24 h.
With the feasibility of catalytic in DMT/[(DMT)H]+ C−H zincation confirmed, we screened several other Brønsted acidic salts. From this, better conversions were achieved using bulkier and more basic amines, e.g., [(Et3N)H]+ salts (entry 3), (see Table S1 for more optimization details). While there was minimal difference switching between two fluorinated aryl-borate anions (entries 3–4, B(ArCF3)4=B{3,5-C6H3(CF3)2}4), more coordinating anions (e.g. [OTf]−) inhibited the catalytic process (entries 5–6). Finally, using two equivalents of heteroarene, and slightly longer reaction times led to the yield of the C−H zincation product 3 a improving to 88 % (entry 7–9). This demonstrates for the first time that C−H zincation catalytic in amine/ammonium salt is feasible and can be high yielding.
Subsequently, we surveyed the scope of this process (Table 2). Replacement of Me with Ph on thiophene led to 3 b forming in 97 % yield. C3-Substituted thiophenes also were amenable, producing 3 c and 3 d. While 3-methyl-thiophene gave both 2-zincated and 5-zincated products (in 16 % and 55 % yield, respectively), the bulkier 3-phenyl derivative produced only the single isomer shown. The more nucleophilic thiophene 3,4-ethylenedioxy-thiophene underwent faster C−H zincation and gave 3 e in 72 % yield within 1 h. 2,2′-Bithiophene and thiophene also underwent regioselective C−H zincation to form 3 f and 3 g in good yield. This zincation methodology also could be applied to furans to provide C−H zincation products 3 h–3 i in moderate yield, with unreacted starting material making up the mass balance. Even the significantly less activated heteroarenes 2-Br-thiophene and benzothiophene (Mayr N value=−2.6)14 were amenable, albeit at a higher temperature for good conversion to 3 j and 3 k. Attempts to C−H zincate N-Me-pyrrole and N-Me-indole did appear to proceed, however, conversions were always <40 % and sufficiently clean samples to permit unambiguous characterization of the products could not be obtained. However, there was no C−H zincation of haloarenes, including those with relatively low pKa values (e.g., C6F5H) in contrast to the Pd-catalyzed process.9 Thus, this C−H zincation is dependent on heteroarene nucleophilicity and not pKa.

- [a] 1 (0.1 mmol), 2 a–k (0.2 mmol), and [(Et3N)H][B(ArCF3)4] (0.01 mmol) in PhCl (0.6 mL) at 60 °C for 24 h unless otherwise stated. Yield by 1H NMR spectroscopy versus CH2Br2 added as an internal standard at the end of the reaction. [b] at 80 °C for 24 h. [c] 16 % zincation at C-2 position also observed. [d] at 60 °C for 1 h. [e] at 80 °C for 48 h. [f] at 100 °C for 24 h.
Next, another NacNac ligand was explored, the smaller N-mesityl derivative, MesNacNacZnH. Using this led to 60 % of the zincation product, 4 (inset bottom Table 2) under the standard conditions. We note that these NacNacZn-Aryl complexes have been shown previously to be effective sources of aryl nucleophiles in Negishi cross-coupling and in allylation reactions.9
The feasibility of using NacNacZnEt in place of zinc hydride 1 also was explored, targeting a “transfer zincation” (Scheme 1) related to the recently reported transfer borylation.15 However, no zincation of 2-methyl-thiophene was observed with either the N-Dipp or N-Mes derivatives (5 a/b) using several ammonium salts. Instead, at ≥120 °C the formation of NacNacZnArF (6 a–d, ArF=C6F5 or 3,5-C6H3(CF3)2) was observed, from decomposition of the anion (see Figure S66–S77). This is likely due to the decomposition of a species such as [NacNacZn−NR3][B(ArF)4] at ≥120 °C as [(R3N)H][B(ArF)4] salts are stable under these conditions.

Attempted C−H zincation with NacNacZnEt.
C−H Alumination Studies
With an effective C−H zincation process in hand we explored the feasibility of extending this approach to another main group metal. Aluminium was selected principally as it is distinct from zinc in multiple ways (e.g., valency and coordination number of the NacNacM−R1/2 complexes). Therefore, if the approach of coupling endergonic C−H metalation with exergonic dehydrocoupling was successful with Al it would indicate general applicability (in terms of metal).
Initial attempts using NacNacAlH2 (7) in place of the zinc-hydride led to the formation of complex intractable mixtures. Therefore, the use of NacNacAlMe2 (8) was explored in place of 7. Pleasingly, the catalytic C−H metalation of 2 a using 8 and 10 mol % [(Et3N)H][B(C6F5)4] proceeded cleanly to give aryl aluminium 9 a as the major product on heating to 80 °C (Table 3 and Table S2 for optimization). Importantly, in the absence of [(Et3N)H][B(C6F5)4] no C−H alumination was observed. This process thus represents the first report of catalytic transfer alumination. In this case the [B(C6F5)4]− anion is essential, with in situ reaction monitoring revealing the formation of several new fluorinated compounds (by 19F NMR spectroscopy) when using [B(ArCF3)4]−. This is consistent with the considerable fluorophilicity of aluminium cations that presumably leads to Al−F bond formation by fluoride abstraction from the sp3-CF3-containing borate anion.

- [a] 8 (0.1 mmol), heteroarene (0.1 mmol), and ammonium salt (0.01 mmol) in PhCl (0.6 mL) at 80 °C for 24 h. Yield by 1H NMR spectroscopy versus CH2Br2 added as an internal standard at the end of the reaction. [b] at 80 °C for 48 h. [c] at 100 °C for 24 h.
Both 2-substituted thiophenes (to form alumination products 9 a and 9 b) and 3-substituted thiophenes (forming 9 c) were amenable to C−H alumination (Table 3). In this case, 3-methyl-thiophene underwent regioselective C−H alumination to provide only the 5-substituted product, 9 c. α-Selective C−H alumination was confirmed for 9 a by single-crystal X-ray diffraction studies (inset top Table 3). This C−H alumination process also was applicable to thiophene, although its lower nucleophilicity17 meant higher reaction temperatures were required. In contrast, the less activated heteroarenes 2-Br-thiophene and benzothiophene did not undergo any C−H alumination at 100 °C. Nevertheless, catalytic C−H alumination worked well for O/N-containing activated heterocycles. For example, furans showed good reactivity (forming 9 e–g), while C−H functionalization of N-TIPS-pyrrole proceeded at the C3-position in moderate yield (46 % 9 h, in 48 h). N-Me-indole underwent selective C−H alumination at the most nucleophilic C3-position to give an excellent (91 %) yield of the product, 9 i. The formation of the C3-metalated isomer 9 i was confirmed by single crystal X-ray diffraction studies (inset bottom Table 3). The C3 functionalization of N-Me-indole under these conditions is consistent with an SEAr-type process and is in contrast to the C−H alumination of N-Me-indole under the previously reported Pd-catalyzed conditions which proceeded selectively at the C-2 position.10 It also should be noted that a range of phenyl derivatives again underwent minimal (or no) C−H alumination as per the zinc system. We also note that it has been shown previously that NacNacAl−(R)Aryl complexes are useful sources of aryl nucleophiles in cross coupling reactions.10c
Next, we sought to convert both the methyl groups in NacNacAlMe2 (8) by increasing the number of equivalents of heteroarene used (from 1 equiv. in the reactions in Table 3). The reaction of 8 with 3 equiv. of 2-methyl-thiophene in the presence of 10 mol % [(Et3N)H][B(C6F5)4] gave 91 % double C−H alumination product 10 after 80 h (Scheme 2).

Double C−H alumination.
This C−H metalation methodology again could be extended to the less sterically demanding MesNacNac ligand. However, in contrast to the zinc system, for aluminium it required a change in the ammonium salt used. Specifically, 73 % C−H alumination of 2-methyl-thiophene with MesNacNacAlMe2 11 (Scheme 3), was achieved to form 12 using 10 mol % [(DMT)H][B(C6F5)4]. In contrast, using 10 mol % [(Et3N)H][B(C6F5)4] and 11/2-methyl-thiophene led to a complex intractable mixture of products.

Double C−H alumination.
Synthesis of Cationic NacNacM Electrophiles and Solid-State Structures
To gain insight into structure–reactivity relationships we synthesized a number of the NacNacZn salts starting from NacNacZnH and [(Base)H][Anion]. This afforded 13 a–e (Figure 2) as single products (by in situ NMR spectroscopy). Given the importance to the catalytic process of forming 13 by dehydrocoupling with [(R3N)H]+ (as the barrier to dehydrocoupling directly impacts ΔG≠span, see below) these reactions were monitored by in situ NMR spectroscopy. This revealed that the more acidic cations, e.g. [(DMT)H]+, resulted in faster dehydrocoupling at room temperature compared to the [(Et3N)H]+ salts, indicating a lower barrier to dehydrocoupling using [(DMT)H]+ (pKa [(DMT)H]+=10.8, [(Et3N)H]+=18.5, both in acetonitrile).16 In contrast to the Zn−H congeners, the NacNacZnEt complexes 5 a/5 b reacted with [(Et3N)H]+ salts only at 50 °C and above, to form 13 c, 13 d (from 5 a) and 13 e (from 5 b), with no reaction observed at room temperature. Thus, there is a higher barrier for ethane formation relative to H2 formation in this system. For details on our studies with other anions (e.g., [(R3N)H]OTf that forms NacNacZnOTf, 14), see Supporting Information S5.9.

A, synthesis of 13 a–e. B, synthesis of 15a–b. C,structures of the cationic components of 13c, 13e and 15a. Ellipsoids at 50 % probability. Hydrogen atoms are omitted for clarity.
The solid-state structures of [NacNacZn(NEt3)][B(ArCF3)4] (13 c) and [MesNacNacZn(NEt3)][B(ArCF3)4] (13 e) were determined (Figure 2C)17 and both found to contain three-coordinate trigonal planar zinc centers with effectively planar (≤0.05 Å deviation) C3N2Zn metallacycles.18 The Zn1−NEt3 bond lengths are similar in 13c and 13e (13c=2.008(4) Å and 13e=1.9945(13) Å) and as expected these Zn←NR3 dative bonds are longer than the covalent bonds in three-coordinate (at Zn) neutral NacNacZn−NR2 complexes (where Zn−NR2≈1.85 Å).19 However, they are shorter compared to those in four-coordinate [NacNacZn(amine)2]+,20 and the TMEDAN−Zn distances in a three coordinate [(TMEDA)Zn−L]+ cation.13f Combined, this data indicates minimal steric destabilization of the Zn−NEt3 bonds in 13 c and 13 e.
Stoichiometric reactions of NacNacAlMe2 with [(Base)H][B(C6F5)4] also were performed. These afforded the cationic aluminium complexes cleanly for base=DMT and Et3N via methane elimination (Figure 2B). The protonolysis reactivity again correlates with the relative Brønsted acidity of the ammonium salts, with 15 a forming within 1 h at room temperature, while the Et3N derivative, 15 b, was formed fully only after heating for a prolonged period at 50 °C. The solid-state structure of [NacNacAl(Me)(DMT)][B(C6F5)4], 15 a (Figure 2C), confirmed the formulation with metrics comparable to other four-coordinate NacNacAl(Me)-based complexes.21, 22 Steric effects in 15 a appear minimal as indicated by the Al−NDMT distance in 15 a (Al1−N3 2.010(6)) being comparable to that in other cationic four-coordinate Al−N(Ph)Me2 complexes that have ligands with less bulky substituents.23 As 15 b did not afford single crystals suitable for X-ray diffraction analysis, the structure of the cationic component was calculated (see below for detailed discussions of computational results). This calculated structure possessed closely comparable metrics to the structure of 15 a indicating minimal differences to the geometry about aluminium on changing the amine from DMT to Et3N.
Mechanistic Studies
Firstly, the endergonic nature of C−H metalation using [NacNacM−NEt3]+ (M=Zn/Al−Me) cations was confirmed. For example, no change in the NMR spectra was observed upon addition of 20 equiv. of 2-methyl-thiophene to the zinc complex 13 c (Scheme 4 top). The subsequent addition of NacNacZnH (1) however still led to the C−H zincation of 2-methyl-thiophene at 60 °C. On complete consumption of 1 during catalytic C−H zincation, the Zn−NEt3 cation 13 c again was observed, suggesting that it is an on-cycle species (Figure S155). Analogous observations were forthcoming from the reaction of the aluminium complex 15 b and 2-methyl-thiophene (Scheme 4, bottom). Consistent with these observations the backwards reactions, i.e. protonolysis of the NacNacM-thienyl complexes 3 a and 9 a with [(Et3N)H]+, proceeded to form 2-methyl-thiophene and 13 c and 15 b, respectively (Scheme 4 and Figure S156–S157).

Endergonic 2-methyl-thiophene metalation.
Next, the kinetic isotope effect (KIE) of deuteration at the C5-position of 2-methyl-thiophene was measured in independent reactions under catalytic conditions. A kH/kD of 1.2 for the C−H zincation of 2-methyl-thiophene was determined. This low value indicates that C−H bond cleavage is not involved in the turnover-limiting step of C−H zincation. Notably, during the catalytic C−H zincation of 5-D-2-methyl-thiophene, 1H and 2H NMR spectra revealed deuterium incorporation into the Cγ position of the NacNac ligand (Figure S159–161). The protonation of the Cγ position of NacNacZnH by [(Et3N)D]+ (formed by deprotonation of 5-D-2-methyl-thiophene during C−D zincation) to form a species such as 16 (Scheme 5) would account for deuterium incorporation. While there is precedence for the Cγ protonation of NacNacM complexes,24 including examples where the Brønsted acid is an ammonium salt,25 it is unprecedented to our knowledge that protonation of the NacNac backbone proceeds in preference to protonation of a M−H unit.

Top, the KIE for C−H zincation of 2-methyl-thiophene. Bottom, proposed protonation of the Cγ position.
As no intermediates were observed during the dehydrocoupling reactions between ammonium cations and NacNacZnH or between NacNacZnEt and [(Et3N)H]+ (see section S7.6–S7.7), definitive evidence for backbone protonation was sought. The reaction of NacNacZnEt with the strong Brønsted acid HNTf2 produced a new compound with a singlet at δ 4.29 in the 1H NMR spectrum, integrating to 2H and assigned to backbone Cγ methylene protons (Figure S162). This is consistent with the formation of compound 17 (Scheme 6), a conclusion supported by the 13C{1H} NMR spectrum containing a resonance for a methylene (γ) carbon at δ 45.6.26 A resonance at δ −77.9 in the 19F NMR spectrum of 17 indicated a non-coordinated NTf2 anion. Combined, this resulted in the formulation of 17 as a three-coordinate at zinc complex, consistent with the observed solution symmetry for the NacNac ligand. Compound 17 converted into 18 at room temperature over time by loss of ethane. The occurrence of backbone protonation prior to dehydrocoupling while notable does not unambiguously confirm that this is an on-cycle process in the C−H zincation. Reversible Cγ protonation followed by direct protonolysis of Zn−H is also feasible, therefore DFT studies were performed to investigate the mechanism further (see below).

The protonation of the Cγ position of 5 a with HNTf2.
For the C−H alumination of 2-methyl-thiophene, a kH/kD of 2.8 was observed based on independent reactions under catalytic conditions (Scheme 7). In contrast to the zinc system, this indicates that a C−H bond cleavage is involved in the turnover-limiting step for C−H alumination. Further contrasts between the Zn/Al systems were forthcoming from analysis of the 1H and 2H NMR spectra which revealed minimal deuterium incorporation into the NacNac Cγ position during C−H alumination even after long reaction times (Figure S180). This suggested a mechanistic divergence in these C−H zincation and C−H alumination reactions.

Determination of C−H alumination KIE.
Finally, to help benchmark the computational studies (see below) an Eyring analysis was performed for the irreversible reaction between NacNacAlMe2 (8) and [(Et3N)H][B(C6F5)4] which forms 15 b and methane (Figure S182–183). This led to a ΔG≠=21.1 (±1) kcal/mol for this step (at 298 K).
Computational Studies on the Mechanism of C−H Zincation and Alumination
The computed profile for the catalytic zincation of 2 a is shown in Figure 3(a), where [NacNacZn(NEt3)]+ (13 c+) is taken as the starting point of the cycle. Overall, the reaction proceeds as postulated, with an endergonic C−H zincation to form NacNacZn(thiophenyl) 3 a and [(Et3N)H]+ (ΔG=+7.7 kcal/mol) being driven forward by exergonic dehydrocoupling with NacNacZnH, 1, to reform 13 c+ with loss of H2 (ΔG=−8.9 kcal/mol). The overall process is therefore exergonic (ΔG=−1.2 kcal/mol). The mechanism of C−H zincation initiates with the associative substitution of NEt3 in 13 c+ by 2-methyl-thiophene via TS1CHZn at +25.9 kcal/mol to form Int1CHZn at +11.6 kcal/mol. Int1CHZn shows elongation of the C4−C5 thiophene bond (1.43 Å cf. 1.38 Å in free 2-methyl-thiophene), consistent with the electrophilic activation of the substrate. Deprotonation by the now free NEt3 base proceeds via TS2CHZn (+24.8 kcal/mol) to give 3 a and [(Et3N)H]+ at +7.7 kcal/mol. Two mechanisms were then characterized for the dehydrocoupling between 1 and [(Et3N)H]+: (i) direct protonolysis of the Zn−H bond, and (ii) a ligand-assisted process. Of these, the latter proved significantly more accessible and proceeds through protonation of the γ-C position of the NacNac backbone via TS1DHCZn at +19.7 kcal/mol to give initially a H-bonded adduct (not shown) from which NEt3 dissociation forms Int1DHCZn. NEt3 coordination at Zn then gives Int2DHCZn at +16.3 kcal/mol. The ZnN2C3 metallacycle in Int2DHCZn (Figure 3b, bottom) exhibits a boat structure with a relatively short C−H⋅⋅⋅H−Zn contact of 2.23 Å. H2 loss then proceeds with a barrier of only 9.2 kcal/mol via TS3DHCZn at +25.5 kcal/mol, that exhibits elongated C−H and Zn−H distances (1.44 Å and 1.79 Å, respectively) and incipient H−H bond formation (C−H⋅⋅⋅H−Zn=1.00 Å). The alternative direct protonolysis of the Zn−H bond in 1 entails a larger barrier of 25.4 kcal/mol via TS4DHCZn at +33.1 kcal/mol (depicted in grey, Figure 3(a) and Figure 3(b) top). For a comparable study with DMT as the base see the Supporting Information (Figure S187).

(a) Computed free energy reaction profile (kcal/mol) for the catalytic C−H zincation of 2-methyl-thiophene (Method: B3PW91(def2-TZVP, D3(BJ), PhCl)//BP86(Zn: SDD; S: SDD(d); other atoms: 6-31G**). a A precursor complex was located at +13.2 kcal/mol; b TS1DHCZn leads initially to a H-bonded NEt3 adduct at +17.9 kcal/mol that is omitted for brevity (see supporting materials). (b) Details of selected stationary points (distances in Å; φ=metallacycle folding angle, see text for details; NacNac Dipp groups, Me H atoms and NEt3 H atoms omitted for clarity).
The computed profile for the C−H alumination of 2 a is shown in Figure 4(a). Starting from the 4-coordinate cationic NEt3 adduct [NacNacAl(Me)(NEt3)]+, 15 b+, the reaction proceeds via the dissociative substitution of NEt3 by 2 a to form the electrophilically activated adduct Int2CHAl at +6.8 kcal/mol. Deprotonation of this species by free NEt3 then gives 9 a, plus [(Et3N)H]+ at +4.2 kcal/mol. This endergonic C−H alumination is followed by an exergonic dehydrocoupling between 8 and [(Et3N)H]+ to reform 15 b+ along with CH4 (ΔG=−15.1 kcal/mol). The overall catalytic cycle is therefore exergonic (ΔG=−10.9 kcal/mol). Calculation of the ligand-assisted protonolysis pathway for Al revealed that while protonation at the NacNac Cγ is kinetically accessible via TS2DHCAl at +19.4 kcal/mol, the subsequent CH4 elimination via TS3DHCAl is much higher at +36.1 kcal/mol. However, in this case direct protonolysis is more accessible via TS1DHCAl (at +24.9 kcal/mol), with this corresponding to a Me-abstraction involving a near-linear Al⋅⋅⋅CH3⋅⋅⋅H⋅⋅⋅NEt3 unit (Figure 4(b) top). Binding of Et3N to Al then occurs in a separate step to reform 15 b+. It is noteworthy that protonolysis (methane loss) and amine binding occur in separate steps in this pathway due to the accessibility of the amine-free cation, [NacNacAl−Me]+ (note, this cation has been previously reported).22 In contrast, in the zinc system forming the amine-free cation, [NacNacZn]+ from 1, results in high energy intermediates (>30 kcal/mol, see Figure S186), precluding a comparable pathway involving direct Zn−H protonolysis followed by a separate amine binding step.

(a) Computed free energy reaction profile (kcal/mol) for the catalytic C−H alumination of 2-methyl-thiophene (Method: B3PW91(def2-TZVP, D3(BJ), PhCl)//BP86(Al/S: SDD(d); other atoms: 6-31G**). a TS2DHCAl leads initially to a H-bonded NEt3 adduct at +17.5 kcal/mol that is omitted for brevity (see supporting materials for details). (b) Details of selected stationary points (distances in Å; φ=metallacycle folding angle, see text for details; NacNac Dipp groups, Me H atoms and NEt3 H atoms omitted for clarity).
The computed reaction profiles in Figures 3 and 4 capture many of the key reactivity patterns seen experimentally. Both C−H zincation and alumination reactions are endergonic and the metalated products are trapped through exergonic dehydrocoupling. The computed barrier for the protonolysis of 1 to form 13 c+ (17.8 kcal/mol) is lower than that for 8 to form 15 b+ (20.7 kcal/mol), consistent with the relative solution reactivities. Furthermore, the computed barrier for the dehydrocoupling of 8 with [(Et3N)H]+ (20.7 kcal/mol) is in good agreement with the experimental value of 21.1 (±1) kcal/mol. The low KIE of 1.2 for the catalytic zincation of 2 a (Scheme 5) is also consistent with the computed rate-limiting transition state, TS1CHZn, that corresponds to associative NEt3/2 a substitution. In contrast, TS3CHAl is computed to be rate-limiting for catalytic alumination and this does involve C−H bond cleavage, aligning with the larger KIE of 2.8 (Scheme 7). More generally, however, both cycles involve several high energy transition states of varying character that are close in energy, and for which the relative energetic ordering is functional dependent. The B3PW91 functional was chosen as it performs qualitatively the best over the range of available experimental mechanistic data; a more quantitative interpretation of the computed profiles would be challenging.
While both the C−H zincation and alumination reactions follow the same general pattern of sequential C−H metalation/dehydrocoupling, these two processes do differ in the details. In some cases, this reflects the valency of the metal centers; NEt3/2 a substitution is an associative substitution at the 3-coordinate Zn in 13 c+, whereas starting from the 4-coordinate Al complex 15 b+ substitution proceeds via a dissociative process. Furthermore, direct protonolysis of the Zn−H bond in 1 involves approach of [(Et3N)H]+ from above the coordination plane in TS4DHCZn, with concerted Zn−H bond cleavage and Zn−NEt3 bond formation. In contrast, such an approach is not found for the direct protonolysis of the Al−Me bond in 8 and instead [(Et3N)H]+ engages in an essentially linear Me abstraction via TS1DHCAl. This is presumably due to the higher coordination number (four in 8 vs. three in 1) and the fact that the amine-free cation [NacNacAl−Me]+ is energetically more accessible than [NacNacZn]+.18, 22
Notably, the barrier to ligand-assisted H2 loss from Int2DHCZn (ΔG≠=9.2 kcal/mol) is much lower than that for ligand-assisted CH4 loss from Int1DHCAl (ΔG≠=21.7 kcal/mol). This significant difference combined with the ubiquity of NacNacM complexes in chemistry27 means the factors impacting the barrier to dehydrocoupling via the ligand assisted pathway warrant further discussion. Two main factors appear to underpin this barrier:
-
H atom transfer is facilitated by the isotropic H 1s orbital that facilitates overlap in the transition state, whereas Me group transfer involves distortion of the more directional sp3 hybrid (as indicated by NBO analyses on these two systems, Figure 5
 Figure 5
Figure 5Coefficients and hybridization of the σZn−H NBO in Int2DHCZn, the σAl−Me NBO in Int1DHCAl and the σγ-C−H NBOs. The impact on orbital overlap in the corresponding transition states is shown schematically (see also Figure S196).
and S196).28 This difference may be enhanced to some extent by the dominant Zn 4s character in the Zn−H bond in Int2DHCZn; in contrast the Al contribution to the Al−Me bond in Int1DHCAl has more directional sp1.2 character. The residual Al⋅⋅⋅Me interaction is further undermined in TS3DHCAl compared to the Zn⋅⋅⋅H interaction in TS3DHCZn.
-
the degree of pre-organization into a boat-like conformation in the precursor complexes, quantified by φ, the metallacycle folding angle between the NMN (M=Zn, Al) and CγCβCβ planes that define the ‘bow’ and ‘stern’ of the boat. In Int2DHCZn φ=91.7°, resulting in a short H⋅⋅⋅H contact of 2.23 Å, and this distortion decreases (φ increases to 97.9°) in TS3DHCZn. In contrast, Int1DHCAl has φ=114.6° (C⋅⋅⋅H=3.16 Å) and the distortion increases to access TS3DHCAl (φ=106.9°). Notably, larger spectator ligands (defined as Y, Table 4
Table 4. Computed barriers (kcal/mol) for loss of H2/MeH from the direct precursor (e.g., Int2DHCZn or Int1DHCAl) and geometric distortions (quantified by the interplane angle φ), in model Zn and Al complexes. Values of φ are for precursor/transition state. 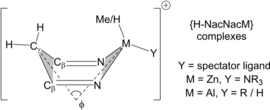
Entry
Precursor Complex
ΔG≠
φ (°)
1
[H−NacNacZnH(NEt3)]+
9.2
91.7/97.9
2
[H−NacNacZnEt(NEt3)]+
13.3
90.6/93.2
3
[H−NacNacAlMe2]+
21.7
114.6/106.9
4
[H−NacNacAlH2]+
16.7
145.7/105.0
5
[H−NacNacAl(H)tBu]+
11.3
102.7/104.2
6
[H−NacNacZnH(NH3)]+
13.9
106.6/100.3
) at the metal promote the ligand-assisted H2 or CH4/ethane loss reactions by increasing the distortion (decreasing φ) of the precursor complexes (see below).

These factors were explored further by computing the ligand-assisted dehydrocoupling step in a series of model complexes (Table 4). [H−NacNacZnH(NEt3)]+ (i.e. Int3DHCZn) and the Et-analogue, [H−NacNacZnEt(NEt3)]+ (Entry 1 vs. 2), show similar φ values, and the higher barrier to ethane loss is consistent with the detrimental directionality effect associated with alkyl transfer. This is consistent with C−H zincation using NacNacZn−Et not being successful due to the larger ΔG≠span produced in part by this effect (see Figure S188). Similarly, CH4 loss from [H−NacNacAlMe2]+ (Int1DHCAl) is harder than H2 loss from the corresponding dihydride [H−NacNacAlH2]+ (Entry 3 vs. 4). Notably, introducing bulky spectator groups to Al (e.g., Entry 5 and Table S10) also increases the distortion (φ=102.7°) and reduces the barrier to H2 loss (see Figure S193) relative to dihydride derivative (Entry 4). A related effect is observed in the zinc system on varying the amine spectator ligand: replacing NEt3 with smaller amines (compare entries 6 and 1, see Table S10 for more examples) decreases the distortion in the precursor (higher φ) and leads to higher barriers to H2 loss.29
It is notable that the kinetically most accessible dehydrocoupling reactions (Entries 1 and 5) involve H2 loss in the presence of the bulkiest spectator ligand explored; the latter pre-organizes the system to such an extent that φ actually increases (i.e., the distortion decreases) in the transition state. This significant effect (up to ca. 5 kcal/mol δΔG≠ in the series studied herein, see Table S10 for more examples) has implications for the design of NacNacM catalysts, as it indicates that increasing the steric bulk around the metal center can actually be desirable for lower barrier protonolysis processes via a ligand-assisted mechanism.
Conclusion
By coupling an endergonic C−H metalation step with a subsequent exergonic dehydrocoupling step we have developed the first catalytic transition metal-free heteroarene C−H zincation/C−H alumination requiring only sub-stoichiometric amine/ammonium salt.
The properties of the base are crucial for an effective process since it fulfils multiple roles: coordination to M and deprotonation of the [M-(heteroaryl-H)]+ complex, with the resultant [(Base)H]+ then key for protonating the Zn−H/Al−Me containing complex. Initial guidelines for selecting an appropriate base include: (i) sufficiently low nucleophilicity towards M to enable displacement by an incoming heteroarene; (ii) sufficient basicity to deprotonate the resultant [M-(heteroaryl-H)]+ complex. Furthermore, combining (i) and (ii) needs to produce a C−H metalation phase that is not too uphill energetically. This is crucial as a significantly endergonic metalation phase will increase the overall energy span of the process once the barrier in the second half of the catalytic cycle, the dehydrocoupling phase, is taken into account, potentially making it too high to proceed. Given that C−H metalation is endergonic, the dehydrocoupling step needs to have a relatively low barrier pathway for the overall cycle to be energetically feasible.
Notably, with NacNac ligands there is a degree of mechanistic flexibility in the M−H/M−Me protonlysis step due to the possibility of direct protonation of the M−H/M−Me unit or ligand non-innocence (backbone protonation) mediating subsequent dehydrocoupling. Computational analysis of both pathways revealed the lowest energy route is predominantly dictated by (i) the accessibility of the amine-free [NacNacM]+ complex and (ii) the degree of steric-derived distortion towards a boat conformer upon backbone protonation. With other ligands backbone protonation may not be feasible, therefore careful selection of the organometallic complex is essential to avoid high energy intermediates/barriers during dehydrocoupling that would therefore preclude catalytic C−H metalation. Based on the insights afforded from this initial study, and the fact it is applicable to two disparate metals (Zn and Al) we believe that multiple other main group metals and other ligand sets will be amenable to this transition metal-free, catalytic C−H metalation approach.
Acknowledgments
This project has received funding from the European Research Council under the European Union's Horizon 2020 research and innovation program (grant agreement No 769599), the University of Edinburgh and the EPSRC (EP/V03829X and EP/T019867/1). We thank the Mass Spectrometry facility (SIRCAMS) at the University of Edinburgh (UoE) for carrying out MS analysis. We also acknowledge Prof. Guy Lloyd-Jones and Dr Dominic Willcox for useful discussions.
Conflict of interests
The authors declare no conflict of interest.

