

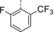
Enantioselective Intramolecular ortho Photocycloaddition Reactions of 2-Acetonaphthones
Graphical Abstract
The intramolecular ortho photocycloaddition of 2-acetonaphthones has been achieved with high enantioselectivity. The catalytic use of a chiral Lewis acid was critical for the success of the reaction and its mode of action has been elucidated by mechanistic experiments and calculations. The primary photocycloaddition products underwent photochemically or thermally a variety of consecutive reactions.

Abstract
2-Acetonaphthones, which bear an alkenyl group tethered to its C1 carbon atom via an oxygen atom, were found to undergo an enantioselective intramolecular ortho photocycloaddition reaction. A chiral oxazaborolidine Lewis acid leads to a bathochromic absorption shift of the substrate and enables an efficient enantioface differentiation. Visible light irradiation (λ=450 nm) triggers the reaction which is tolerant of various groups at almost any position except carbon atom C8 (16 examples, 53–99 % yield, 80–97 % ee). Consecutive reactions were explored including a sensitized rearrangement to tetrahydrobiphenylenes, which occurred with full retention of configuration. Evidence was collected that the catalytic photocycloaddition occurs via triplet intermediates, and the binding mode of the acetonaphthone to the chiral Lewis acid was elucidated by DFT calculations.
Introduction
In recent years, there has been a rapidly increasing number of studies which aimed to employ arenes and hetarenes as substrates in ortho photocycloaddition reactions.1 In the course of this transformation, an olefin adds either inter- or intramolecularly to a formal double bond of the aromatic core, leading to a rupture of the aromatic π system (dearomatization2). While the reaction is comparably facile at five-membered heterocycles with a pronounced double bond character,3 benzenoid arenes are more difficult to address and often require high energy UV photons (λ≤380 nm) for the reaction to occur.4 For benzene and its derivatives, the intermediately formed bicyclo[4.2.0]octa-2,4-diene ring system is frequently unstable, and consecutive processes occur which have been exploited for the synthesis of complex carbocyclic scaffolds.5, 6 For condensed benzenoid arenes, such as naphthalene7 and phenanthrene,8 the respective ortho photocycloaddition products are thermally stable and can be isolated. The reaction can be executed by direct excitation or by addition of a sensitizer. Electron withdrawing groups at the arene shift the wavelength bathochromically and ensure an improved regioselectivity in the addition process.
The study presented in this manuscript aimed at an enantioselective ortho photocycloaddition reaction from naphthalenes to cyclobutanes, for which there is so far no precedence.9 One key discovery that initiated the current project was the finding that Lewis acids induce a bathochromic shift in aromatic aldehydes such as phenanthrene-9-carboxaldehyde (1) which in turn facilitates their excitation at long wavelength (Scheme 1).10

The ortho photocycloaddition of phenanthrene-9-carboxaldehyde (1) can be performed enantioselectively upon irradiation at λ=457 nm in the presence of chiral Lewis acid 2 a (top).10 The intramolecular ortho photocycloaddition of 1-substituted 2-acetonaphthones 4 delivers racemic cyclobutanes rac-5 (bottom).11 The reaction can also be conducted at long wavelength upon sensitization.12
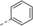
Subsequent addition reactions occur at the C9−C10 double bond and deliver cyclobutanes such as 3 in an ortho photocycloaddition reaction. The transformation can be performed enantioselectively13 upon proper choice of a Lewis acidic catalyst. In this context, AlBr3-activated oxazaborolidines have been found to be an excellent choice14 and complex 2 a enabled the transformation 1→3 with high enantioselectivity (ee=enantiomeric excess).
In search for reactions which allow for the formation of complex cyclobutanes from benzenoid arenes, we were intrigued by the intramolecular ortho photocycloaddition of 2-acetonaphthones 4 which display a pendant olefin linked to carbon atom C1.11 The reaction to racemic cyclobutanes rac-5 can be performed either at short wavelength (pyrex filter, λ >280 nm) by direct excitation or with visible light by energy transfer.12 We hypothesized that the reaction might be performed enantioselectively, provided that a Lewis acid would enable an excitation beyond the absorption wavelength of the uncomplexed substrate—either in the presence of a sensitizer15 or potentially even in its absence. A major challenge of this endeavor was the mode of binding to the Lewis acid. Chiral oxazaborolidines had so far been reported to deliver a high enantioselectivity in photochemical reactions, only if the substrate offered—apart from the Lewis basic carbonyl oxygen atom—a suitable hydrogen atom for a second non-classical binding to the oxygen atom of the oxazaborolidine. The required hydrogen atom was either the aldehyde hydrogen atom as present in substrate 1 or an α-hydrogen atom in α,β-unsaturated carbonyl compounds.14 In recent work on the thermal, oxazaborolidine-catalyzed enantioselective Diels–Alder reaction of 2,3-disubstituted cyclobutenones,16 we have found that α,β-unsaturated enones lacking an α-hydrogen atom can deliver high enantioselectivities. This discovery spurred hope that the substitution pattern around the carbonyl group17 might also be expanded for enantioselective photochemical reactions. In fact, the present study has now revealed that an acetyl group in 2-acetonaphthone provides a suitable handle for Lewis acid coordination and a highly enantioselective ortho photocycloaddition of the title compounds was developed. The synthetic study was accompanied by quantum chemical calculations of the Lewis acid-substrate complex and additional mechanistic studies. Full details of our work are disclosed in this research article.
Results and Discussion
Optimization and Scope. Preliminary experiments on a potentially enantioselective ortho photocycloaddition were performed with substrate 4 a. For comparison, racemic product rac-5 a was obtained by irradiation (λ=450 nm) of naphthone 4 a in the presence of 1 mol % of [Ir(ppy)2(dtbbpy)](PF)6 (ppy=2-(2-pyridinyl)phenyl; dtbbpy=4,4′-di-tert-butyl-2,2-dipyridyl).12a In the optimization reactions, yield and ee of product 5 a were monitored employing activated oxazaborolidines with different substituents Ar and Ar1 (Table 1). The oxazaborolidines were prepared in situ from the respective proline-derived amino alcohol (variation of Ar) and a boronic acid (variation of Ar1). AlBr3 was subsequently added to generate the putative complexes 2 (for details, see the Supporting Information). Although it was not clear whether it would be required, the iridium complex [Ir(Fppy)2(dtbbpy)](PF6) with a reported triplet energy of ET=223 kJ mol−1 (CH2Cl2, r.t.)15b was added (Fppy=4-fluoro-2-(2-pyridinyl)phenyl) in the first set of screening experiments. The reaction was performed at −78 °C to avoid further skeletal rearrangements of product 5 a (see below). Light-emitting diodes (LEDs) with an emission range centered at λ=450 nm served as light sources. Initial results were somewhat disappointing (2 b–2 e, entries 1–4) as either the yield or the enantioselectivity remained low. However, the beneficial effect of a 3,5-disubstitution at the phenyl groups of the amino alcohol (Ar) became already apparent (entry 3). A breakthrough was achieved with a 2,6-disubstitution at the aryl substituent at the boronic acid (Ar1). Even with a relatively small phenyl group at the amino alcohol, the enantioselectivity increased to 72 % ee for Ar1=2,6-dichlorophenyl (2 f, entry 5) and to 86 % ee for Ar1=2-fluoro-6-trifluoromethylphenyl (2 g, entry 6). Further optimization was conducted by altering the Ar group in the prolinol backbone (entries 7–9), and the best result (86 % yield and 89 % ee) was obtained using Lewis acid 2 j (entry 9). Likely reasons for the excellent performance of catalysts 2 h–2 j are their electron deficient aryl substituent Ar1 (increasing the Lewis acidity) and the steric bulk of their aryl groups Ar (facilitating an efficient enantioface differentiation).
|
|||||
entry[a] |
Ar |
Ar1 |
cat. |
yield[b] [%] |
ee[c] [%] |
|---|---|---|---|---|---|
1 |
|
|
2 b |
52 |
38 |
2 |
|
|
2 c |
19 |
39 |
3 |
|
|
2 d |
13 |
52 |
4 |
|
|
2 e |
3 |
36 |
5 |
|
|
2 f |
76 |
72 |
6 |
|
|
2 g |
78 |
86 |
7[d] |
|
|
ent-2 h |
76 |
−84 |
8 |
|
|
2 i |
85 |
87 |
9 |
|
|
2 j |
86 |
89 |
10[e] |
|
|
2 j |
71 |
92 |
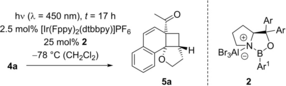
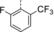
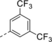
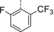
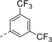
- [a] Test reactions were performed on a scale of 0.2 mmol. [b] Yield of isolated product. [c] Determined by chiral HPLC analysis. [d] The (R) enantiomer of the catalyst was used. [e] The reaction was performed without iridium complex.
While the optimization went on, we studied in parallel the UV/Vis-properties of the Lewis acid complex formed from 2-acetonaphthone 4 a (see below). Although EtAlCl2 was the Lewis acid employed in the spectroscopic study, the results indicated that the Lewis acid complex might be competent to react in the absence of the iridium catalyst at λ=450 nm. In fact, under these conditions (entry 10), the desired product was obtained in 71 % with an improved enantioselectivity of 92 % ee. The result marks to the best of our knowledge the first enantioselective photocycloaddition of unsaturated ketones mediated by a chiral oxazaborolidine catalyst. The high efficiency of the reaction indicated that the iridium catalyst had not been required in most of the other screening reactions (Table 1, entries 1–9) but they were not repeated. Instead, the optimized conditions (λ=450 nm, 25 mol % 2 j, −78 °C, CH2Cl2) were applied to other 2-acetonaphthones 4 with a tethered olefin (Scheme 2). Further lowering the catalyst loading led to a decrease in yield and enantioselectivity (see the Supporting Information for details).

Enantioselective ortho photocycloaddition of 1-substituted 2-acetonaphthones 4 as catalyzed by Lewis acid 2 j (0.2 mmol scale) [a] 63 % yield, 90 % ee (1 mmol scale). [b] Addition of [Ir(dFppy)2(dtbbpy)](PF6) (2.5 mol %). [c] Lewis acid 2 g was used. [d] The reaction was performed at −50 °C. [e] Addition of [Ir(dF(CH3)ppy)2(dtbbpy)](PF6) (2.5 mol %). [f] Lewis acid ent-2 h was used.
Substrates 4 with electron withdrawing substituents, including methoxycarbonyl (4 b), fluoro (4 c), chloro (4 d), and bromo (4 e) at position C7 were amenable to the optimized reaction conditions, affording products 5 b–5 e smoothly in 84–93 % yields and 92–93 % ee. The absolute configuration of product 5 e was unambiguously determined by single crystal X-ray diffraction analysis (anomalous diffraction)18 and the configuration assignment for all other products was based on analogy. Surprisingly, naphthyl ketone 4 f with an electron-rich methyl group gave a low conversion under optimal conditions. In this instance, both yield and enantioselectivity were improved when triplet sensitizer [Ir(dFppy)2(dtbbpy)](PF6) was added (dFppy=3,5-difluoro-2-(2-pyridinyl)phenyl; see Tables S1–2 for more details). Substrates 4 g and 4 h with substituents at positions C5 and C6 were readily processed, giving products 5 g–5 h in 85–93 % yield and 94–95 % ee. The reaction of substrates 4 i–4 j with substituents at position C4 was preferentially performed with catalyst 2 g delivering the desired products in 87–90 % ee. In the former case, an iridium complex was added as co-catalyst to improve the yield. The reaction of 4 k with a bromo substituent at position C8 was sluggish, probably due to steric hindrance. The reaction was promoted by elevating the temperature slightly, and the product 5 k was obtained in 23 % yield and 60 % ee at −50 °C. The reaction of 6,7-disubstituted but-3-enyl-1-oxyacetonaphthones (4 l and 4 m) provided the corresponding products (5 l and 5 m) smoothly in 70–99 % yield and with 80–92 % ee. In the latter instance, [Ir(dF(CH3)ppy)2(dtbbpy)]PF6 [dF(CH3)ppy=2-(2′,4′-difluorophenyl)-5-methylpyridine] was utilized to improve both yield and enantioselectivity (see Table S3 for optimization). Apart from substrates substituted at the arene core, alkenes were surveyed which bear substituents at the olefinic double bond. Remarkably, a substitution was tolerated at the internal carbon atom (substrate 4 n), at one external position (substrates 4 o, 4 p) and at both external positions (substrate 4 q). Employing catalyst ent-2 h, cycloaddition products ent-5 n–5 q were smoothly obtained in good yields and with a high degree of diastereoselectivity for ent-5 o and ent-5 p. The enantioselectivities were in the range of 88–97 % ee.
Apart from the fact that compounds 5 and ent-5 represent complex three-dimensional scaffolds for applications in synthesis and drug discovery,19 they also offered the option of a conversion to another cyclobutane-containing motif. Glorius and co-workers had reported that a sensitized irradiation of the compounds initiates a rearrangement reaction to tetrahydrobiphenylenes.12a The putative reaction course as envisioned for photocycloaddition product 5 a involves a cleavage of the internal cyclobutane bond in the triplet state T1 (Scheme 3).

Suggested reaction pathway for the photochemical rearrangement of product 5 a to tetrahydrobiphenylene 7 a (≙: equivalent to).12a Energy transfer leads to a cleavage from the respective triplet (T1) 1,4-diradical 6 a which undergoes ring closure to the product.
The resulting 1,4-diradical 6 a can assess several reaction pathways one resulting in product 7 a by C−C bond formation. Although product 7 a appears to be the preferred product under photolytic conditions and although the reaction appears to be not reversible, it remained unclear whether the absolute configuration was retained in the process. In fact, triplet 5 a (T1) cannot only form 1,4-diradical 6 a but can also undergo cleavage to the starting material 4 a. Since the ensuing ortho photocycloaddition would be unselective in this instance, the enantiomeric excess would deteriorate. Our concerns were manifested by initial experiments performed with iridium complex [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [dF(CF3)ppy=2-(2′,4′-difluorophenyl)-5-trifluoromethylpyridine] as the sensitizer (Scheme 4). The latter compound can initiate the desired rearrangement but it is, due its triplet energy of ET=255 kJ mol−1 (MeCN, r.t.),20 also competent to catalyze the reaction 4 a→rac-5 a in an unselective fashion. Irradiation of substrate 5 a (90 % ee) resulted in the formation of product 7 a with a diminished enantiomeric purity of only 77 % ee. Recovered (recd.) starting material 5 a was also partially racemized suggesting that the ortho photocycloaddition was reversible. Gratifyingly, the photolytic cleavage could be suppressed at lower temperature and it was found that already at T=0 °C the chirality transfer was close to complete (Scheme 4a).

Skeletal rearrangement of cyclobutanes 5 to products 7 upon sensitized irradiation[a] Sensitizer (sens): [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. The chirality transfer is more efficient at low temperature (a) and deuterium incorporation remains complete (b). Several substrates were converted in moderate to good yields to the rearranged products (c) when irradiated at 0 °C or −20 °C (7 h, 7 l) for 14–18 h in acetonitrile solution (for details see the Supporting Information).
Deuteration experiments (Scheme 4b) supported the hypothesis that the loss in enantioselectivity was due to reversible C−C bond fission. No deuterium scrambling was observed indicating that the C−H bond at the stereogenic center was not cleaved.
With the optimized conditions for the skeletal rearrangement established, the protocol was applied to a selection of ortho photocycloaddition products 5 (Scheme 4c). If the reaction temperature was kept at 0 °C, the chirality transfer was high as corroborated by the formation of products 7 b, 7 g, and 7 i. For some substrates, the reactions could be performed at even lower temperature (−20 °C), and tetrahydrobiphenylenes 7 h and 7 l were isolated in high enantiomeric purity (93–95 % ee). The enantioselective ortho photocycloaddition, thus, gives not only access to tetracyclic products 5 with a lateral cyclobutane but also to consecutive products 7 with an internal four-membered ring.
The two most obvious synthetic handles for a further functionalization of products 5 are the exocyclic acetyl group and the olefinic double bond within the six-membered ring. Two exemplary reaction were performed on compound 5 a to demonstrate the synthetic utility of the photocycloaddition products (Scheme 5). Dihydroxylation with stoichiometric quantities of N-methylmorpholine N-oxide (NMO) gave diol 8 in 47 % yield as a single diastereomer.21 The front face of the olefin is exposed to the attack of the reagent while the acetyl group and the tetrahydrofuran ring block the back face. The relative configuration of product 8 was established by single crystal X-ray crystallography.22 As a prototypical transformation of the acetyl group in compound 5 a, triflate 9 (Tf=trifluoromethanesulfonyl) was formed upon deprotonation with lithium diisopropylamide (LDA) and subsequently subjected to a Negishi cross-coupling23 reaction that delivered olefin 10.

Consecutive reactions of photocycloaddition product 5 a (top) and of rearrangement product 7 h (below). Both compounds were employed in high ee (92 % and 95 %) and there was no erosion of the relative and absolute configuration in the described reactions (Cy=cyclohexyl).
Likewise, tetrahydrobiphenylene 7 h served as a representative substrate for consecutive reactions of this substrate class. A diastereoselective cuprate addition24 delivered saturated ketone 11, which was then subjected to a Suzuki cross-coupling reaction.25 The structure of final product 12 was corroborated by single crystal X-ray crystallography.26
Mechanistic studies and computational results. In preliminary experiments, we studied the absorption properties of 2-acetonaphthone 4 a in the presence of varying amounts of the strong Lewis acid EtAlCl2 (Figure 1). The UV/Vis spectrum of compound 4 a resembles parent 2-acetonaphthone, for which spectroscopic data have been reported previously. Reported absorption maxima at λ=245 nm (ϵ=70000 M−1 cm−1), at λ=280 nm (ϵ=10000 M−1 cm−1), and at λ=335 nm (ϵ=2000 M−1 cm−1)27 correlate well with the maxima found for compound 4 a at λ=245 nm (ϵ=33100 M−1 cm−1), at λ=287 nm (ϵ=7600 M−1 cm−1), and at λ=334 nm (ϵ=2500 M−1 cm−1).

UV/Vis spectrum of 2-acetonaphthone 4 a (c=1.0 mm in CH2Cl2, quartz cuvette, ø=1.0 mm) in the presence of variable equivalents (eq.) of EtAlCl2.
It is assumed that the nπ* transition of 2-acetonaphthone is hidden by the strong ππ* transition at long wavelength. Upon addition of increasing amounts of the Lewis acid, an absorption at long wavelength (λmax=416 nm) evolves which increases gradually. Isosbestic points at λ=258 nm, 284 nm, and 300 nm indicate that the complex is a 1 : 1 complex with no other species being involved. In agreement with UV/Vis studies on the complex between parent 2-acetonaphthone and Mg(ClO4)2 as the Lewis acid,28 we assume that the long wavelength absorption results from a red shift of the ππ* transition thus activating the chromophore for irradiation with visible light (λ>380 nm).
In previous work on the intramolecular [2+2] photocycloaddition of α,β-unsaturated carbonyl compounds, it was observed that the uncatalyzed reaction is more efficient than the reaction under Lewis acid catalysis. Although the absorption coefficient of the nπ* transition was low (ϵ≤100 M−1 cm−1), the quantum yield Φ of the photocycloaddition was high and was found for the specific case of 1-(pent-4-enoyl)-2,3-dihydropyridin-4(1H)-one (13) to exceed 0.23 (λ=366 nm, CH2Cl2, −70 °C).29 In the presence of a Lewis acid (50 mol %), the quantum yield was determined under otherwise identical conditions as Φ=3.8×10−3 (Scheme 6).

Quantum yields Φ determined in previous work29 for the [2+2] photocycloaddition 13→14 in the absence and in the presence of a Lewis acid.
The loss in efficiency was accounted for by the fact that, according to El-Sayed's rules,30 intersystem crossing (ISC) from an nπ* singlet to a ππ* triplet is allowed but forbidden for states with ππ* character. Since the lowest lying triplet (T1) is responsible for the photocycloaddition in both cases, with and without Lewis acid, and since it is ππ* in character, the uncatalyzed reaction benefits from the high ISC rate and can successfully compete with the catalytic reaction. As a consequence, high chiral Lewis acid loadings (50 mol %) were required to guarantee high enantioselectivities in photochemical processes which occur in the triplet manifold.29, 31
In the present case, we had seen that the catalyst loading can be lowered to 25 mol % without compromising the enantioselectivity in the reaction and we wondered whether this observation would also be reflected in the quantum yields. The parent acetonaphthone 4 a was employed for the experiments and it was first converted to racemic ortho photocycloaddition product rac-5 a upon direct irradiation at λ=366 nm (Scheme 7). The quantum yield was found to be Φ=0.13 indicating that the reaction is efficient but that other decay pathways exist apart from the photocycloaddition reaction (see below). Since the oxazaborolidine Lewis acids are not stable at ambient temperature, the catalytic reaction was performed with EtAlCl2 (25 mol %) as the Lewis acid. Given the observed bathochromic shift and given the irradiation conditions for the enantioselective reaction, the wavelength was switched to λ=424 nm. Under these conditions, the quantum yield was determined as Φ=1.1×10−2 confirming a—compared to the reaction 13→14—more efficient reaction under Lewis acid catalysis. In the absence of Lewis acid, there was no conversion at λ=424 nm.

Quantum yields Φ determined for the ortho photocycloaddition 4 a→rac-5 a in the absence and in the presence of a Lewis acid.
It is well established for naphthyl-substituted carbonyl compounds, that their triplet state is rapidly (<1 ns) populated by direct excitation and ISC.32 Thus, the uncatalyzed reaction 4 a→rac-5 a with a quantum yield of Φ=0.13 is very likely to operate via the corresponding triplet excited state. Given the analogy to the enone [2+2] photocycloaddition (see above), it is tempting to assume that also the reaction in the presence of the Lewis acid proceeds via triplet intermediates. While the fact that sensitization with iridium catalyst was in several cases successful (products 5 f, 5 i, 5 m) supports the triplet hypothesis, additional experiments were performed to substantiate the involvement of triplet state intermediates.
Due to its low triplet energy, 1,3-pentadiene (piperylene) is known to quench triplet intermediates efficiently and has been exploited extensively as a mechanistic tool in photochemistry.33 We studied the progress of the reaction 4 a→5a in the presence of chiral Lewis acid 2 j (cf. Scheme 1) at −78 °C in dichloromethane solution. The conversion, i.e. the ratio of starting material (sm) or product (p) to the total concentration [Σ(sm+p)], was monitored over time (Figure 2). In the absence of the quencher the reaction progressed rapidly and reached a conversion of ca. 65 % after five hours. Upon addition of five equivalents of piperylene under otherwise identical conditions, the reaction rate decreased significantly reaching a conversion of only 30 % after five hours. A further decrease in reaction rate was noted upon addition of ten equivalents of piperylene (ca. 20 % conversion).

Quenching experiments with chiral Lewis acid 2 j (25 mol %) and varying equivalents (eq.) of 1,3-pentadiene (piperylene) as triplet quencher (sm=starting material; p=product).
It has been shown for the Lewis acid-catalyzed oxadi-π-methane rearrangement reaction, that the addition of piperylene does not interfere with a chiral AlBr3-activated oxazaborolidine,34 ruling out the possibility of catalyst poisoning being responsible for the rate decrease. The qualitative result of a rate decrease upon piperylene addition can consequently be considered as circumstantial evidence for a triplet pathway. The same set of quenching experiments was performed for the uncatalyzed reaction 4 a→rac-5 a at λ=366 nm which is a triplet process. The qualitative results were identical (see the Supporting Information for details) with a significant rate decrease upon addition of five or ten equivalents of piperylene.
A second set of experiment supporting the hypothesis of a triplet pathway was performed with olefins 4 o as substrates for the enantioselective ortho photocycloaddition (Schemes 2 and 8). A stereoconvergent reaction course typically indicates the existence of a species in which a rotation around the former C=C double bond is possible.35 In other words, if (E)- and (Z)-olefin give the same product diastereoisomer in a photocycloaddition, a triplet pathway is likely responsible.

Stereoconvergent formation of product ent-5 o occurs both from (E)-4 o (cf. Scheme 2) and from (Z)-4 o. The trans configuration in product ent-5 o supports a reaction course, in which triplet 1,4-diradical 15 is involved.
The reaction of substrate (E)-4 o gave product ent-5 o (Scheme 2) in which the alkyl chain and the methoxy carbonyl group are trans positioned. While this outcome would be consistent with a concerted pathway, the fact the (Z)-isomer (Z)-4 o delivers predominantly the same diastereoisomer (Scheme 8, L.A.=Lewis acid) indicates the formation of a triplet intermediate, most likely 1,4-diradical 15.
The absolute configuration of the ortho photocycloaddition was determined by X-ray diffraction studies (see above). Since we were curious about the factors responsible for the stereochemical outcome, we performed calculations on the 1 : 1 complex of substrate 4 a and Lewis acid 2 j. Complex C1 mimics a situation in which the major enantiomer 5 a of the reaction is formed by attack at the carbon atom in 1-position from the Si face. Complex C2 simulates an attack at the Re face which would lead to the minor enantiomer ent-5 a. The dihedral angle between the C=O double bond and the arene double bond is denominated as angle ϕ and defined as shown in Figure 3.

Two diastereomeric complexes C1 and C2 form upon coordination of substrate 4 a to Lewis acid 2 j. In complex C1, the olefin of the but-3-enyloxy group approaches the arene double bond from the Si face, in complex C2 from the opposite Re face. The angle ϕ defines the dihedral angle between the C=O double bond and the arene double bond.
We investigated the conformational space of the complex formed by substrate 4 a with catalyst 2 j by manually rotating the dihedral angle ϕ (see Figure 3) in constrained gas-phase density functional theory (DFT) structure optimizations using the PBE density functional36 with D3 dispersion correction37 and the def2-SVP basis set38 in the Q-Chem electronic-structure package.39 By rotating over the full 360° both clockwise and counterclockwise, we identified seven local minima (see the Supporting Information for structures)—four for C1 (Si face attack) and three for C2 (Re face attack). Rotation in both directions was necessary due to the flexibility of the olefin arm. No additional low-lying conformers were identified in a metadynamics conformer analysis performed with CREST40 and the semiempirical density functional tight-binding method GFN2-xTB.41 The minimum structure for complex C2 is stabilized by a hydrogen bond between the substrate and the catalyst (see Figure 4b). This is true also for the most stable structure for complex C1, that is additionally stabilized by π-π interaction between the naphthyl unit in 4 a and one aryl group of the catalyst (see Figure 4a). The latter structure is also the global minimum accounting for the enantioselectivity of the reaction. The relative energetic ordering did not change when solvent effects were included with a polarizable continuum model (PCM)42 considering a dielectric constant of ϵ=8.93 for dichloromethane. To arrive at accurate free energy differences, we devised a composite approach with three additive terms for i) the electronic energy, ii) a correction for the solvation free energy and iii) a free energy correction using the harmonic oscillator/rigid rotor approximation. To obtain the vacuum electronic energy, we first re-optimized the two lowest-energy structures with the more accurate ωB97X-V functional43 in the presence of the PCM solvent model using the same def2-SVP basis set.

Ball-and-stick representation of the C1 minimum structure (4a) and the C2 minimum structure (4b). C atoms are colored black, H atoms are white, O atoms are red, N is blue, F atoms are yellow, B is purple, Br is green, and Al is silver. Hydrogen bonds are visualized as green dotted lines. The π-interacting naphthyl and aryl units in (4a) are highlighted with blue face filling.
Under this assumption, we calculate a very high enantiomeric excess of ≥99 % ee, which corresponds well with the experimentally observed result. Since the preference for C1 over C2 seems to be controlled by dispersion interactions in the form of π–π interactions, we repeated the calculations with the ωB97X-D3 functional.48 We consistently found a preference for the complex C1 but with a slightly smaller free energy difference between C1 and C2 minimum structures (10.6 kJ mol−1). We also analyzed the effect of different solvent models (see Supporting Information) without any significant alteration of the result.
Combining mechanistic studies and calculations, a coherent picture for the enantioselective reaction of substrates 4 evolves. Lewis acid coordination not only leads to a significant bathochromic shift of the absorption but also entails a high enantioface differentiation. The methyl group of the acetyl substituent is involved in a favorable hydrogen bonding interaction, which in concert with π-stacking directs the attack of the internal olefin to the Si face of the naphthalene core (relative to carbon atom C1). Previous calculations49 suggest that the propensity for the favored product controlled by the S0 thermodynamics is further amplified by the excited-state topology of the triplet surface. The ensuing triplet 1,4-diradical is competent of cyclobutane ring formation but can also lead to starting materials after ISC. It is likely that a sterically unsuited tetrahydrofuran formation (trans but cis) is corrected by retro cleavage, which in turn lowers the quantum yield. The final products 5 are stable under the reaction conditions and are readily displaced by substrate from the catalyst given their considerably larger size.
Conclusion
In summary, it has been discovered that an intramolecular ortho photocycloaddition of 2-acetonaphthones can be performed with high enantioselectivity. Up to four consecutive stereogenic centers with a defined configuration are created in a single operation. From a synthetic point of view, it is particularly noteworthy that the primary cyclobutanes 5 obtained from the reaction are stable compounds, which underwent distinct consecutive transformations. The reaction is the first example for a methyl ketone being successfully employed in an enantioselective photochemical reaction mediated by a chiral Lewis acid. The hydrogen bonding interaction, as corroborated by DFT calculations (structure C1, Figure 4), may be a useful tool to design future photochemical reactions of ketones. From a mechanistic perspective, a bathochromic shift, as observed for compounds 4 upon Lewis acid coordination (chromophore activation), is responsible for the fact that the reaction can be performed by visible light irradiation in the absence of a triplet sensitizer. The Lewis acid-catalyzed reaction is more efficient than previous enantioselective enone [2+2] photocycloaddition reactions and can be performed with a catalyst loading of only 25 mol %. A triplet reaction pathway was secured for the reaction 4→5 (quenching experiments, stereoconvergent reaction), and also the course of the consecutive photochemical reaction 5→7 could be elucidated. A reversible C−C bond cleavage was identified as key element of the rearrangement which led to enantiomerically enriched tetrahydrobiphenylenes 7 with perfect chirality transfer.
Acknowledgments
PY, QW, and PL are grateful to the National Key R&D Program of China (2022YFA1503200); National Natural Science Foundation of China (21921003) for financial support. SS and TB acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—TRR 325 (project B1)—444632635 for financial support. EK and CJS received financial support by the DFG under Germany's Excellence Strategy—EXC 2089/1-390776260 (e-conversion). Computations were carried out using computational and data resources provided by the Leibniz Supercomputing Centre (www.lrz.de). We thank J. Kudermann (TU München) for his help with GLC analyses and Dr. S. Breitenlechner and J. Hofer (both TU München) for their help with the quantum yield determination. Open Access funding enabled and organized by Projekt DEAL.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

