Radical Coupling Initiated by Organophosphine Addition to Ynoates
Graphical Abstract
Combining established two electron nucleophilic catalysis of organophosphines with photoredox organocatalysis has allowed the discovery of a mechanistically unusual Giese coupling reaction. In addition to addressing a limitation in traditional photoredox coupling with electron-poor alkynes these studies demonstrate the viability of reaction designs that merge traditional nucleophilic phosphines organocatalysis and photoredox events.

Abstract
Dual nucleophilic phosphine photoredox catalysis is yet to be developed due to facile oxidation of the phosphine organocatalyst to the phosphoranyl radical cation. Herein, we report a reaction design that avoids this event and exploits traditional nucleophilic phosphine organocatalysis with photoredox catalysis to allow the Giese coupling with ynoates. The approach has good generality, while its mechanism is supported by cyclic voltametric, Stern–Volmer quenching, and interception studies.
Partnering nucleophilic organocatalysis with photoredox processes has enabled the discovery of unique radical reactions.1 Although various organocatalysts have been exploited in such designs, organophosphines remain undeveloped. This deficiency is due to the ready oxidation of organophosphines to the phosphoranyl radical cation (i.e. 1, Figure 1A). This species, known for some time,2 undergoes umpolung coupling to nucleophiles (to give 2) before undergoing various fragmentations to enable novel reactions.3 For example, in Zhu's pioneering work triphenyl phosphine is used stoichiometrically to give radical 2 a that ultimately allows acyl radical coupling to alkenes (Figure 1C).3a In a related vein, recent studies from Shi exploit oxidation of quinuclidine to its amino radical cation which allowed radical-radical coupling of electronically neutral and rich olefins to phtalimides (Figure 1D).4 However, contrasting such studies the addition of organophosphines as a Lewis base to electrophiles, a ubiquitous event in phosphine organocatalysis,5 is yet to be merged with photoredox events in a dual catalytic reaction (Figure 1B). If such dual catalysis is viable then this introduces the possibility to leverage the diverse chemistry of nucleophilic phosphine organocatalysis5 in novel radical coupling reactions.

Background. A) Oxidation of organophosphines to the phosphoranyl radical cation 1 is facile. B) Conceptual background. C) Zhu's, acyl radical coupling via 2 a. D) Shi's quinuclidine catalyzed coupling. E) Summary of reaction reported herein.
To demonstrate this concept, we considered a dual catalytic approach to the Giese reaction of electron-poor alkynes.6 Although photoredox approaches to the Giese chemistry of alkenes are well developed,7 couplings to alkynes remain challenging8, 9 with reactions of electron-poor alkynes even more limited.10 Conceptually we envisaged exploiting the established polarity inversion of ynoates (i.e. 5) to give phosphonium species 3 a11 in the two electron phosphine organocatalytic stage. Based on phosphonium reagent photoredox chemistry established by McNally,12c we expect that this catalytically generated species should be more easily reduced than the ynonate, thereby allowing reductive coupling with trifluoroborate 6 to ultimately give the Giese product 7 (Figure 1E). Herein, we report the realization of this concept which both introduces a unique Giese reaction, and highlights that conventional nucleophilic phosphines can be successfully merged with photoredox events.
Studies commenced by examining the reduction potential of alkyne 5 b and phosphonium 3 b. Cyclic voltammetry reveals an increase by more than 1 volt confirming that the phosphonium is more easily reduced than the alkyne (Figure 2A). Next, attention was directed to the radical coupling partner. A radical precursor with a lower oxidation potential than triphenyl phosphine (E1/2=+0.98 V vs SCE)2a was targeted to avoid phosphoranyl radical cation formation (Figure 1A). While benzyl trifluoroborate salt 6 a (R1=R2=H, R3=Ph; E1/2=+1.1 V vs SCE)13 has an oxidation potential greater than Ph3P, the 2° variant 6 b is lower (E1/2=+0.89 V vs SCE) and potentially suited to the proposed design (Figure 2A). Thus, a detailed mechanistic hypothesis was conceived in which polarity inversion of alkyne 5 b would give vinyl phosphonium 3 b, while a suitably reducing excited state photocatalyst (PC*) would then provide zwitterionic radical 4 b and the oxidized photocatalyst (PC⋅+) (Figure 2B). Next alkyl radical 7 b, formed by oxidation of the radical precursor 6 b hence regenerating the photocatalyst (PC), would undergo radical-radical coupling to provide 9 b before elimination of the triphenyl phosphine would give the product 7 b.

Reaction design and discovery. A) Oxidation and reduction potentials for key intermediates and catalysts. B) Mechanistic design. C) Reaction discovery, optimizations and control experiments.
Discovery and optimization commenced by identifying a suitable photocatalyst. Screening a range of photocatalysts, in the presence of 20 mol % PPh3, and sodium acetate/acetic acid buffer11d in acetonitrile, demonstrated that PC114 gave the cross-coupled product 7 b in 68 % yield (Figure 2C, entry 1). Due to low solubility, the photophysical properties of this catalyst in acetonitrile are unknown, however data in dichloromethane14 is consistent with the proposed design. Namely, reduction of the vinyl phosphonium 3 b by the excited state photocatalyst (E1/2 (PC*/PC⋅+)=−0.99 V vs SCE) and oxidation of the trifluoroborate 6 b by the oxidized photocatalyst (E1/2 (PC⋅+/PC)=+1.44 V vs SCE). Interestingly, the alternative scenario in which oxidation of the trifluoroborate 6 b by the excited state catalyst (E1/2 (PC*/PC⋅−)=+1.41 V vs SCE) and reduction of the vinyl phosphonium 3 b by the reduced photocatalyst (E1/2 (PC⋅−/PC)=−1.02 V vs SCE) to give the zwitterionic radical 4 b provides an equally plausible mechanistic explanation. The reaction was also viable with PC2-PC5 however the yield decreased by 19 to 30 % with these catalysts (Figure 2C, entries 2–5). Performing the reaction without photocatalyst gave phosphonium 3 b in 72 % yield (based on PPh3) with no coupled product (Figure 2C, entry 6). Using less Lewis basic phosphines, the yield was reduced, while the more Lewis basic, and presumably more easily oxidized P(4-MeOC6H4)3 was not suited to the reaction (Figure 2C, entries 7–9). In the absence of the phosphine catalyst, or light source, the reaction failed (Figure 2C, entries 10 and 11), while removing the buffer led to a decrease in yield from 68 % to 26 % (Figure 2C, entry 12).
Studies into the reactions generality demonstrated the successful union of 3° and 2° radicals with alkynes conjugated to esters (Table 1, entries 1–13) or aliphatic ketones (Table 1, entries 14–18). Some sensitivity to electronics about the aryl group of the trifluoroborate was observed, such that the 2-FC6H5 product 7 f formed in 32 % yield (Table 1, entry 5). This decrease in yield is likely due to challenges with the chemoselective oxidation revealed by CV studies that showed a 0.1 V increase in 6 f (E1/2=+0.99 V vs SCE) compared to benzyl 6 b. Replacement of the methyl group by ethyl or i-propyl was possible (Table 1, entries 7 and 8) as was the use of alternate esters (Table 1, entries 11 and 12). While the less readily oxidized 6 a (R1=Ph, R2=R3=H)13 discussed previously could not be coupled using the standard conditions when the less readily oxidized phosphine P(4-ClC6H4)3 was used a 13 % yield of the expected product was isolated (Table 1, entry 13). Alkyl enones were well tolerated with products containing saturated heterocycles (Table 1, entries 14 and 15), strained rings (Table 1, entry 17) as well as simple chains (Table 1, entry 16) and rings (Table 1, entry 18) formed effectively.

- [a] Isolated yield, [b] Using P(p-ClC6H4)3 rather than PPh3, [c] Isolated as a 5 : 1 mixture of diastereomers.
Mechanistic studies commenced by examining the role of the phosphonium intermediate 3. Using vinyl phosphonium bromide 3 b and subjecting it to the reaction conditions the expected product 7 b formed in 78 % or 75 % yield, with or without additional phosphine, confirming its viability as an intermediate (Figure 3A). While 13P NMR monitoring gave unreliable results during irradiation, it was possible to identify phosphonium 3 b prior to irradiation, with no free triphenylphosphine (see Supporting Information for details). The facile formation of 3 b is likely to also play a role in avoiding the oxidation of triphenyl phosphine.

Mechanistic studies. A) Phosphonium bromide 3 b can produce the cross-coupled product 7 b. B) Stern–Volmer studies demonstrate quenching of excited state PC1 (4CzIPN) by vinyl phosphonium 3 b and BF3K salt 6 b in acetonitrile. C) TEMPO trapping studies with ynoate 5 b and phosphonium 3 b are consistent with formation of zwitterionic radical 4 b. D) Lack of cyclized material 11 is consistent with radical-radical coupling.
Steady-state luminescence quenching experiments (Stern–Volmer studies) were not possible with the PC1 catalyst due to low emission intensities, however the PC2 catalyst (E1/2 (PC*/PC⋅+)=−1.04 V vs SCE), which was viable in the reaction could be analyzed. These studies confirmed that the phosphorescence intensity from the excited-state photocatalyst PC2*, was quenched in the presence of either vinyl phosphonium bromide 3 b or BF3K salt 6 b with the expected linear Stern–Volmer plot (Figure 3B). These studies demonstrated quenching rate constants (Kq) of 1.84×104 M−1s−1 and 1.18×104 M−1s−1, for 3 b and 6 b, respectively. The higher observed rate for 3 b is consistent with the proposed mechanism (Figure 2B), however based on reaction stoichiometry, and catalyst loading of Ph3P, both previously introduced mechanisms are plausible. To distinguish between these mechanisms at the very least the use of the correct catalyst would be required.
Next we examined the formation of zwitterionic radical 4 b using TEMPO trapping studies. Thus, when the reaction was performed with 4 equivalents of TEMPO the trapping product 10 b was isolated in 69 % yield. While the benzylic radical was not identified, significant quantities of its dimer were. It was also possible to demonstrate that the phosphonium 3 b can provide zwitterionic radical 4 b. Thus, repeating this reaction with phosphonium 3 b gave TEMPO adduct 10 b in 79 % isolated yield (Figure 3C).
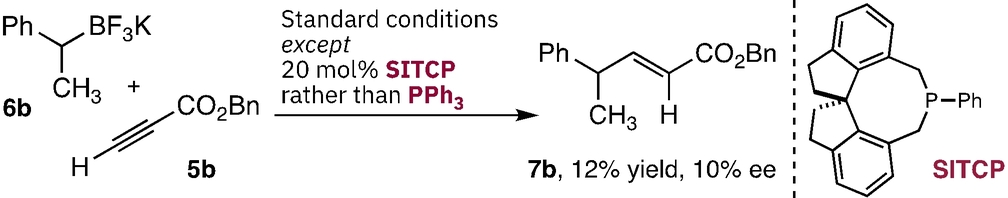
Further support for a radical-radical coupling mechanism involving 4 a could be obtained by considering the cross-coupling reactions of BF3K salt 6 b with ynoates 5 t–v. Each ynoate contains a tethered alkene capable of facile 5-exo-trig cyclization to give increasingly stable radicals.15 This process might be expected if direct radical addition to either the phosphonium 3 b or the ynoate 5 b is occurring. When examined, each gave the regular cross-coupled materials 7 t–v without any cyclization products (11) (Figure 3D). This result, along with TEMPO trapping studies, and literature precedent with stoichiometric couplings of pyridyl phosphoniums,12c provides support for the radical-radical fragment coupling pathway. Finally, preliminary studies into an enantioselective variant of this reaction have been undertaken with a range of chiral phosphines. While most proved unsuitable, SITCP provided the coupled product 7 b in 12 % yield and 10 % ee. This result highlights a potential approach to this reaction, although clearly distinct catalysts with greater reactivity, and stereoselectivity, are required.16
The nucleophilic addition of phosphines to electron-poor π-systems is the bedrock of much of phosphine organocatalysis.5 The absence of reports on dual catalysis with photoredox events is at first surprising. However, our studies highlight that reaction conditions to avoid oxidation of the highly Lewis basic, and reducing, catalyst to the phosphoranyl radical cation2, 3, 4 must be judiciously identified, and that this provides boundaries for reaction design. Specifically, in the chemistry reported herein the use of radical precursors more readily oxidized than triphenyl phosphine was essential, with electron-rich catalysts not viable (Figure 1C, entry 9). However, with considered reaction design discovery is indeed possible. In the future, we envisage that thoughtfully designed reactions should allow the rich annulative chemistry of phosphine organocatalysis involving allenoates, MBH adducts, and related species, to potentially deliver radical coupling-reactions with even greater ability to deliver molecular complexity.
Acknowledgments
The authors thank the Australian Research Council through the Discovery (DP210103530) programs for financial support. Open Access publishing facilitated by Monash University, as part of the Wiley - Monash University agreement via the Council of Australian University Librarians.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.