

Arylation of Axially Chiral Phosphorothioate Salts by Dinuclear PdI Catalysis
Graphical Abstract
The first direct C−SP(=O)(OR′)(OR′′) coupling of diverse and chiral phosphorothioate salts with aryl iodides, enabled by an air- and moisture-stable PdI dimer is demonstrated. The mechanistic and computational data suggest distinct dinuclear PdI catalysis to be operative, which allows for operationally simple couplings with broad scope and full retention of stereochemistry.

Abstract
S-aryl phosphorothioates are privileged motifs in pharmaceuticals, agrochemicals, and catalysts; yet, the challenge of devising a straightforward synthetic route to enantioenriched S-aryl phosphorothioates has remained unsolved to date. We demonstrate herein the first direct C−SP(=O)(OR′)(OR′′) coupling of diverse and chiral phosphorothioate salts with aryl iodides, enabled by an air- and moisture-stable PdI dimer. Our mechanistic and computational data suggest distinct dinuclear PdI catalysis to be operative, which allows for operationally simple couplings with broad scope and full retention of stereochemistry.
While phosphorothioates display privileged structure/activity features and find applications as important agrochemicals, top-selling pharmaceuticals, and powerful chiral catalysts (see Scheme 1 A), the control of their inherent chirality remained a daunting synthetic challenge for decades. Indeed, indirect multistep strategies via PIII species have historically been utilized to accomplish this feat,1 until Baran's recent announcement of accessing chiral phosphorothioate oligonucleotides by an indirect strategy from limonene derived PV reagents under auxiliary control (Scheme 1 C).2 On the other hand, the synthesis of chiral phosphorothioate salts has been realized through a straightforward three step sequence.3 As such, if it would be possible to further derivatize these salts at sulfur, without compromising their stereochemical integrity, then access to the wider phosphorothioate class of compounds would in principle be unleashed.

Motivation and synthetic strategy.
In this context, the synthesis of chiral aryl phosphorothioates, which are commonly found in drugs, pesticides, and bioactive compounds,4 would require the direct coupling of the salt with a Csp2 center (Scheme 1 D). If realizable, this strategy would be highly powerful, as it would allow for the introduction of the phosphorothioate moiety in any desired chirality and at any stage in a synthesis. However, although thiolations of Csp2 sites, especially aryl halides, are well precedented and can be accomplished by Pd0-catalyzed cross coupling reactions,5 to date, no general, safe, robust and direct Csp2–SP(=O)(OR2)(OR3) coupling to access aryl phosphorothioates exists.6 Instead, the vast majority of synthetic approaches rely on a bond formation at the phosphorus center directly (S–P coupling, Scheme 1 B),7 which has so far not been accomplished in a stereoselective manner.
We therefore set out to develop a method to access diverse and chiral phosphorothioates in a direct coupling approach with widely available aryl halides.
Owing to the general mildness and high functional group tolerance, which are key requirements for late-stage synthetic applications, a Pd0/PdII catalyzed cross-coupling approach appears ideal at first sight to accomplish this goal. However, when we subjected various Pd0 catalysts to iodobenzene and NMe4[SP(=O)(OPh)2],8 such as Pd(PPh3)4 or Pd2(dba)3/dppf, no coupling was observed (see Figure 1 A). Moreover, when we subjected the phosphorothioate salt to a PdII complex and heated the corresponding mixture, we did not observe the corresponding product 2.10 Our computational studies support this observation,9 indicating that the iodide/phosphorothioate exchange at [LnPd(II)(Ph)(I)], followed by reductive elimination of Ar−SP(=O)(OPh)2 has a prohibitively high activation free energy barrier of ΔG≠≥30 kcal mol−1 and is endergonic. For comparison, the corresponding thiolation, that is, Ar−SPh reductive elimination is characterized by about 10 kcal mol−1 lower activation free energy barrier and significantly increased driving force (see Supporting Information for details). As such, and in agreement with the lack of literature precedence, Pd0/PdII catalysis appears to not be an optimal strategy to accomplish C−SP(=O)(OR2)(OR3) coupling.

Free energy profile of PdI-catalyzed coupling of PhI with [SP(O)(OPh)2]−. Gibbs free energies (in kcal mol−1) shown, calculated at the SMD (toluene) M06L/def2TZVP//B3LYP-D3/6-31G(d, p) (SDD for Pd, I) level of theory.
We previously established that dinuclear PdI catalysis is a viable strategy to circumvent potentially poisonous PdII intermediates as well as alter the driving force of the transformation.11 Our extensive mechanistic studies had indicated that the bridging iodides of the dimer are initially exchanged by the nucleophilic coupling partner, and the newly formed PdI complex then reacts with the aryl halide, therefore formally reversing the sequence of elementary steps (Figure 1 B). As such, we envisioned that a dinuclear PdI coupling protocol11, 12 might offer a solution to the phosphorothioate problem.
To assess whether PdI-dimer catalyzed phosphorothioation would be feasible, we undertook computational studies of the coupling with iodobenzene, which clearly indicated that there is a pronounced driving force to convert iodobenzene to the C−SP(=O)(OPh)2 coupled product (Figure 1). The activation barrier was found to be in the range of our previous findings of dinuclear PdI catalysis,11, 12 and as such appeared to be feasible.
A key requirement of the PdI dimer catalysis concept is that the coupling partner will also function as a stabilizing bridging unit in the PdI dimer. To examine this, we subjected 4 equiv. of tetramethyl ammonium phosphorothioate salt to a solution of the air-stable, iodide bridged PdI dimer 1. To our delight, within 2 hours at room temperature we saw the appearance of a signal at 97.9 ppm upon 31P-NMR spectroscopic analysis, which is very reminiscent of a functionalized PdI dimer, suggesting that the iodide in 1 can be exchanged for a phosphorothioate.13
We next set out to explore whether PdI catalyzed phosphorothioation of aryl iodides is also feasible. To our delight, the desired O,O-diphenyl phosphorothioate 214 was obtained in 83 % yield in the presence of 5 mol % of [PdI(μ-I)(PtBu3)]2 dimer 1 (Table 1). Encouraged by these findings, we subsequently turned to explore the wider scope of the transformation (Table 1). A series of aryl iodides bearing electron-donating or electron-withdrawing substituents (R=4-Me, 4-nBu, 4-tBu, 4-F, 4-Cl and 4-Ph) reacted smoothly and gave the corresponding products 3–8 in good yields. The reactions of sterically hindered ortho-substituted (2-MeO, 2-Me, and 2-Ph) aryl iodides also proceeded well, with slightly lower yield observed with meta substitution (9–12). For 10, prolonging the reaction time increased the product yield. The reactions of disubstituted substrates were also successful without any loss of yield (13 and 14). This was also true for the polyaromatic iodides giving rise to products 15 and 16. Moreover, the method tolerated a variety of functional groups, such as esters and morpholines (17–19).
|
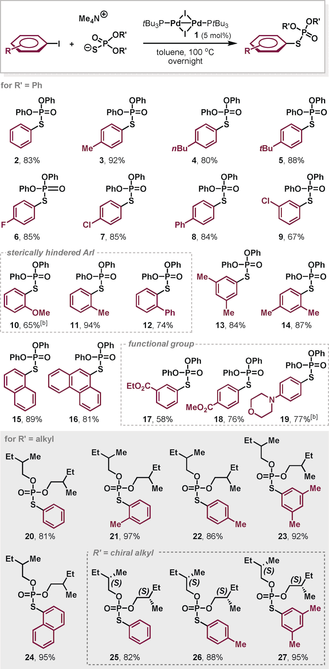
- [a] Yields of isolated products 2–27 after chromatography. [b] The reaction mixture was stirred for 36 hours.
The effect of O,O-dialkyl phosphorothioate salt was examined as well. Various substituted aryl iodides were readily coupled under the standard reaction conditions and the corresponding products 20–24 were obtained in excellent yields. Notably, the chirality in the substituents of the salt was retained (25–27).
The PV-series of TADDOL derived organophosphorus compounds are usually found in various valuable chiral catalysts and ligands.15 To further expand the synthetic utility of the present catalytic strategy, the synthesis of axially chiral S-aryl phosphorothioates was tested (Table 2). Gratifyingly, uniformly excellent enantioselectivities of up to >99 % ee were obtained using C2-symmetric TADDOL-derived phosphorothioate salts with a variety of aryl iodides, resulting in the desired products 28–39 in very good yields. The absolute configuration of the product 30 was determined by X-ray crystallographic analysis (Table 2), unambiguously confirming the complete retention of chirality.16
|
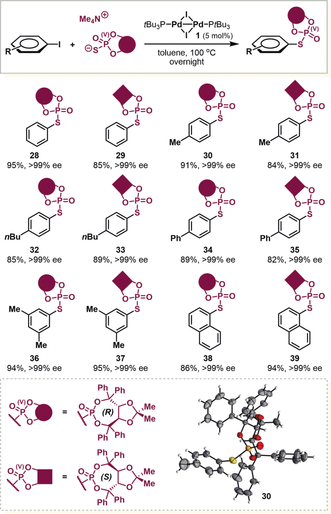
- [a] Yields of isolated products 28–39 after chromatography and the ee determined by HPLC analysis of the purified product on a chiral stationary phase.
In conclusion, while the examined Pd0/PdII catalysis failed to deliver the phosphorothioation of aryl iodides as a consequence of disfavored exchange at PdII and a high reductive elimination barrier, the mechanistically distinct dinuclear PdI catalysis operates with an orthogonal driving force that allows for the first catalytic S-arylation of phosphorothioate salts with aryl iodides. The method is characterized by generality, broad scope, and operational simplicity, making use of an easily accessible phosphorothioate salt and an air-stable PdI dimer. The stereochemical integrity of chiral salts was not compromised in the coupling process, allowing the efficient assembly of axially chiral S-aryl phosphorothioates. In light of the promising properties of S-aryl phosphorothioate compounds as well as the privileged PdI catalysis reactivity, we anticipate that the presented methodology will facilitate numerous applications in the agrochemical, pharmaceutical, and stereoselective synthesis arenas.
Acknowledgements
We thank the RWTH Aachen and the European Research Council (ERC-637993) for funding. Calculations were performed with computing resources granted by JARA-HPC from RWTH Aachen University under project “jara0091”.
Conflict of interest
The authors declare no conflict of interest.

