

Genetic analysis of Parkin in early onset Parkinson's disease (PD): Novel intron 9 g > a single nucleotide polymorphism and risk of Taiwanese PD†‡
Chun-Hsien Wu and Chih-Ying Chao contributed equally to this work.
How to Cite this Article: Wu Y-R, Wu C-H, Chao C-Y, Kuan C-C, Zhang W-L, Wang C-K, Chang C-Y, Chang Y-C, Lee-Chen G-J, Chen C-M. 2010. Genetic Analysis of Parkin in Early Onset Parkinson's Disease (PD): Novel Intron 9 g > a Single Nucleotide Polymorphism and Risk of Taiwanese PD. Am J Med Genet Part B 153B:229–234.
Abstract
Early onset Parkinson's disease (PD) has been associated with mutations in Parkin. We screened Parkin mutations in a cohort of Taiwanese early onset PD using direct cDNA sequencing. Two deletions (Ex2-3del and Ex5del), one point mutation (R334C), one 86-bp IVS9 insertion (c.1084intron+), and two polymorphisms (S167N and V380L) were identified. The mutations identified are heterozygous and none of the mutation carriers possess two Parkin mutations. The c.1084intron+ was due to a novel IVS9 g > a change. To assess the association of IVS9 g > a, S167N and V380L with the risk of PD, we conducted a case–control study in a cohort of PD and ethnically matched controls. Although the difference is not significant, the V380L C allele frequency was notably lower in PD patients than the controls and a trend toward decrease in risk of developing PD was evident (odds ratio: 0.71, 95% confidence interval: 0.53–0.97, P = 0.029). Contrarily the IVS9 g > a a allele frequency was notably higher in PD patients than the controls and a trend toward increase in risk of developing PD was also evident (odds ratio: 1.65, 95% confidence interval: 1.06–2.59, P = 0.028). Quantitative real-time PCR showed that the relative Parkin c.1084intron+ mRNA expression was increased in PD patients with IVS9 ga genotype as compared to gg genotype. Pairwise genotype analysis revealed that IVS9 gg genotype strengthens the negative association of the V380L GC genotype with PD (odds ratio: 0.67, 95% confidence interval: 0.48–0.94, P = 0.021). The results of Parkin mutation/polymorphism screening may contribute to our understanding of PD. © 2009 Wiley-Liss, Inc.
INTRODUCTION
Parkinson's disease (PD) is a common neurodegenerative disorder caused by loss of dopaminergic neurons in the brain's nigrostriatal pathway [Lang and Lozano, 1998]. Clinical features of PD include resting tremor, bradykinesia, rigidity, and postural instability. The etiology of PD has not been fully elucidated. An interaction between environmental factors and genetic predisposition, most of which are not yet known, are thought to cause PD [Eriksen et al., 2005]. Causal mutations of SNCA, Parkin, PINK1, DJ-1, LRRK2, and ATP13A2 genes have been identified mostly in early onset PD and rarely in late-onset PD [reviewed by Belin and Westerlund, 2008]. Genetic mutations in most sporadic cases of PD are rare and the primary cause of the majority of PD remains unknown.
The Parkin gene was initially identified in several families with a rare autosomal recessive juvenile parkinsonism [Kitada et al., 1998]. In addition to functioning in the ubiquitin-mediated proteosomal degradation system [Giasson and Lee, 2003], Parkin is also involved in the regulation of mitochondrial transcription/replication [Kuroda et al., 2006]. The frequency of Parkin mutations was estimated to be as much as 50% in autosomal recessive juvenile parkinsonism [Lücking et al., 2000], 10–25% in early onset PD with age at onset (AAO) between 20 and 50 years of age [Hedrich et al., 2002; Kann et al., 2002; Poorkaj et al., 2004] and 2–6% in late-onset PD [Klein et al., 2003; Oliveira et al., 2003a]. A wide variety of mutations including exon deletions, duplications, rearrangements, point mutations, and splice site mutations have been observed in a variety of ethnic groups (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PARK2). Exonic deletions/duplications of Parkin are generally de novo, whereas common European founders seem to explain the missense mutations discovered in Europe and the United States [Periquet et al., 2001; Lincoln et al., 2003]. Various Parkin mutations were also reported in early onset Taiwanese PD [Lu et al., 2001; Wu et al., 2002, 2005; Shyu et al., 2005]. In addition to the reported mutations, exonic S167N and V380L single nucleotide polymorphisms (SNPs) in the Parkin gene were identified [Wang et al., 1999].
To evaluate the contribution of Parkin gene to the development of sporadic PD in Taiwan, we performed Parkin cDNA sequencing for 55 early onset PD patients, determined heterozygous deletion/insertion by real-time quantitative PCR analysis, and analyzed mutations and polymorphisms in a large sample of patients and controls.
MATERIALS AND METHODS
Subjects
A total of 506 unrelated Taiwanese PD subjects (44.1% females) were recruited from the neurology clinics of Chang Gung Medical Center. All patients were diagnosed with probable idiopathic PD according to the published criteria by two neurologists specialized in movement disorders (Y.-R. Wu and C.-M. Chen) [Gelb et al., 1999]. They all exhibit at least three of four cardinal signs of PD including resting tremor, cogwheel rigidity, bradykinesia or asymmetric onset and have sustained response to levodopa or a dopamine agonist. Subjects with atypical presentations or prior history of multiple cerebrovascular events or other causes of parkinsonian symptoms (e.g., brain injury or tumor, encephalitis, antipsychotic medication) were excluded. The mean age at onset of PD was 62.8 ± 11.1 years, ranging between 19 and 86 years (10.9% of PD patients had onset before age 50). A group of 508 normal control individuals without neurodegenerative diseases were recruited from the same ethnic community. Control subjects (45.7% females) had mean age at examination of 60.5 ± 13.4 years, ranging between 22 and 94 years. All examinations were performed after obtaining informed consent from patients and control individuals.
Mutation/Polymorphism Screening
DNA was extracted from leucocytes using the standard protocols. For PD patients with onset ≤50 (n = 55), RNA was extracted using PAXgene Blood RNA Kit (PreAnalytiX, Qiagen, Hilden, Germany). The RNA was DNase (Stratagene, La Jolla, CA) treated, quantified, and reverse-transcribed to cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The Parkin cDNA was polymerase chain reaction (PCR) amplified (Table I), gel purified and sequenced directly using the ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). The reported R334C and the novel c.1084intron+ were verified by genomic DNA PCR and sequencing. Allele-specific PCR was designed for R334C population screening (Table I). For case–control studies, the S167N (rs1801474), V380L (rs1801582) and IVS9 g > a (novel) SNPs were determined using the AlwNI (loss of site), Bsp1286I (gain of site) and XhoI (loss of site) restriction enzymes, respectively (Table I).
| Test (amplified region) | Anneal (°C) /MgCl2 (mM) | Product/RFLP enzyme (fragment, bp) |
|---|---|---|
| cDNA sequencing (exon 1–7) | ||
| F: CCACCTACCCAGTGACC | 54/1.5 | 827 |
| R: TGTCACACAGTATAAGTGGAA | ||
| cDNA sequencing (exon 6–12) | ||
| F: CGCAACAAATAGTCGGAACA | 58/1.5 | 729 |
| R: GCGCCCGGCCGCCCTGG | ||
| S167N (AGC > AAC) RFLP (exon 4) | ||
| F: ACAAGCTTTTAAAGAGTTTCTTGT | 52/1.0 | AlwNI: CAGN3CTG (167, 94/261) |
| R: AGGCAATGTGTTAGTACACA | ||
| V380L (GTA > CTA) RFLP (exon 10) | ||
| F: ATTGCCAAATGCAACCTAATGTC | 52/1.0 | Bsp1286I: GDGCHC (165/107, 58) |
| R: TTGGAGGAATGAGTAGGGCATT | ||
| IVS9 (g > a) RFLP (intron 9) | ||
| F: TGATCCACCCGCCTTGG | 56/1.5 | XhoI: CTCGAG (199, 101/300) |
| R: AGCAAAAGAGAGGTTCTCTTCCC | ||
| R334C (CGC > TGC) allele-specific PCR (exon 9) | ||
| F: GGCTGAAATTTGCAGTCAGT | 55/1.5 | 278/176 |
| F3: GGGGCGTGTTATGCCCCT | ||
| R: AATATAATCCCAGCCCATGTGCA | ||
- The underlines in the primer sequence and enzyme recognition site indicate the allele-specific nucleotide and polymorphic site, respectively.
Parkin Expression Analysis
To verify Ex2-3del and Ex5del, real-time quantitative PCR was performed on a leucocyte cDNA amount equivalent to 100 ng total RNA with TaqMan fluorogenic probes Hs01038318_m1 (exon 1–2 boundary), Hs01038327_m1 (exon 4–5 boundary), and Hs00247755_m1 (exon 8–9 boundary) for Parkin and 4326321E for HPRT1 (endogenous control) (Applied Biosystems). Expression level was calculated using the formula 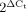 , ΔCt = Ct (HPRT1) − Ct (Parkin). The amount of c.1084intron+ mRNA was determined by customized Assays-by-Design probe (Forward primer: GAGCCTGACCAGAGGAAAGTC, Reverse primer: CAGTGCACAGATGAGGAAATCG, TaqMan® probe: ACTGACCCCACAGCCC) (Applied Biosystems). Relative c.1084intron+ mRNA expression level was calculated using the formula
, ΔCt = Ct (HPRT1) − Ct (Parkin). The amount of c.1084intron+ mRNA was determined by customized Assays-by-Design probe (Forward primer: GAGCCTGACCAGAGGAAAGTC, Reverse primer: CAGTGCACAGATGAGGAAATCG, TaqMan® probe: ACTGACCCCACAGCCC) (Applied Biosystems). Relative c.1084intron+ mRNA expression level was calculated using the formula 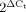 , ΔCt = Ct (Parkin) − Ct (c.1084intron+).
, ΔCt = Ct (Parkin) − Ct (c.1084intron+).
Statistical Analysis
The genotype frequency data and the expected genotypic frequency under random mating were computed and Chi-square tested for Hardy–Weinberg equilibrium using standardized formula. The genotype association analysis was carried out using the Chi-square test. The SNPSpD method [Nyholt, 2004] was used to generate an adjusted significance threshold for correction of multiple SNP testing. The experiment-wide significance threshold of 0.017 was required to keep the type I error rate at 5%. Odds ratios with 95% confidence intervals are calculated to test association between the genotype/allele and disease. Statistical analysis of c.1084intron+ mRNA differences between the groups was carried out using one-way analysis of variance (ANOVA).
RESULTS
Mutation/Variant Analysis of Parkin
Parkin cDNA fragments from 55 early onset PD patients were amplified for sequence analysis. Three reported (R334C, Ex2-3del, and Ex5del) [Hattori et al., 1998; Lücking et al., 2000; Biswas et al., 2006] and a novel c.1084intron+ (86 or 90-bp insertion) mutations were identified (Fig. 1A). The mutations identified are heterozygous and none of the mutation carriers possess two Parkin mutations. The R334C mutation was only found in this early onset PD patient and not found upon screening 452 control chromosomes.

Mutation identification. A: cDNA sequence analysis of R334C, Ex2-3del, Ex5del, and c.1084intron+ mutations. Sequences located in exon are indicated by uppercase letters and in intron by lowercase letters. B: DNA sequence analysis of IVS9 g > a polymorphism. C: Restriction enzyme analysis of IVS9 g > a. The PCR amplified products were digested with XhoI and resolved on a 2% agarose gel. Individual with gg genotype demonstrates complete digestion of the 300-bp PCR product into products of 199 and 101 bp (lane 2), whereas individual with aa genotype demonstrates only the 300-bp product (lane 3). Individual with ga genotype has all three products (lane 1). Lane M (HinfI digest of pGEM4 DNA) is the size marker. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
For the novel c.1084intron+, in silico searches revealed that c.1084 intron+ was flanked by cryptic splice acceptor (splice site strength score 5.795) and donor (splice site strength score 9.762) sites within IVS9; the regular IVS9 acceptor and IVS10 donor sites have scores of 9.629 and 11.150, respectively (http://es.embnet.org/∼mwang/assp.html). Sequence analysis of region flanking the insertion revealed that the c.1084intron+ may result from a novel g > a transition change located at position -6 of the cryptic splice acceptor site (Fig. 1B), which may slightly increase the use of the cryptic splice acceptor site (from score 5.795 to score 6.092). The IVS9 g > a change is polymorphic which can be differentiated using the XhoI restriction analysis (Fig. 1C).
Case–Control Study of S167N, V380L, and IVS9 g > a
As IVS9 g > a may increase the chance of false splicing of 86-bp IVS9 leading to c.1084intron+, we conducted a case–control study in a cohort of PD (n = 506) and ethnically matched controls (n = 508) to assess the association of IVS9 g > a and the reported S167N (AGC > AAC in exon 4) and V380L (GTA > CTA in exon 10) [Wang et al., 1999] with the risk of PD. The genotype and allele distributions of the three SNPs in patients and controls are displayed in Table II. All genotype frequencies confirmed to be in the Hardy–Weinberg equilibrium. There was no statistically significant difference (P > 0.017) in genotype or allele distribution between patients and controls for all three SNPs examined, after correction of multiple SNP testing. However, for V380L, the frequency of C allele (7.9% vs. 10.7%, P = 0.037) was notably lower in PD patients than the controls. When odds ratios of the at-risk genotype/allele were calculated, V380L C allele demonstrated a trend toward decrease in risk of developing PD (odds ratio: 0.71, 95% confidence interval: 0.53–0.97, P = 0.029). On the contrary, the IVS9 g > a a allele was notably higher in PD patients than the controls (5.2% vs. 3.2%, P = 0.030), and a trend toward increase in risk of developing PD was evident (odds ratio: 1.65, 95% confidence interval: 1.06–2.59, P = 0.028). Pairwise genotype analysis revealed that IVS9 gg genotype slightly strengthens the negative association of the V380L GC genotype with PD (odds ratio: 0.67, 95% confidence interval: 0.48-0.94, P = 0.021).
| Genotype/allele | PD | Control | Odds ratioa (95% CI) | P-value | ||
|---|---|---|---|---|---|---|
| No. | % | No. | % | |||
| S167N G > A | ||||||
| GG | 183 | 36.2 | 186 | 36.6 | 1.00 | |
| GA | 252 | 49.8 | 235 | 46.3 | 1.09 (0.83–1.43) | 0.533 |
| AA | 71 | 14.0 | 87 | 17.1 | 0.83 (0.57–1.20) | 0.327 |
| G | 618 | 61.1 | 607 | 59.7 | 1.00 | |
| A | 394 | 38.9 | 409 | 40.3 | 0.95 (0.79–1.13) | 0.543 |
| V380L G > C | ||||||
| GG | 427 | 84.4 | 403 | 79.3 | 1.00 | |
| GC | 78 | 15.4 | 101 | 19.9 | 0.73 (0.53–1.01) | 0.057 |
| CC | 1 | 0.2 | 4 | 0.8 | 0.24 (0.01–1.60) | 0.197 |
| G | 932 | 92.1 | 907 | 89.3 | 1.00 | |
| C | 80 | 7.9 | 109 | 10.7 | 0.71 (0.53–0.97) | 0.029 |
| IVS9 g > a | ||||||
| gg | 457 | 90.3 | 475 | 93.5 | 1.00 | |
| ga | 45 | 8.9 | 33 | 6.5 | 1.42 (0.89–2.28) | 0.143 |
| aa | 4 | 0.8 | 0 | 0.0 | — | |
| g | 959 | 94.8 | 983 | 96.8 | 1.00 | |
| a | 53 | 5.2 | 33 | 3.2 | 1.65 (1.06–2.59) | 0.028 |
| V380L/IVS9 g > a | ||||||
| GG/gg | 387 | 76.5 | 373 | 73.4 | 1.00 | |
| GG/ga | 37 | 7.3 | 30 | 5.9 | 1.19 (0.72–1.98) | 0.500 |
| GG/aa | 3 | 0.6 | 0 | 0.0 | — | |
| GC/gg | 69 | 13.6 | 99 | 19.5 | 0.67 (0.48–0.94) | 0.021 |
| GC/ga | 8 | 1.6 | 2 | 0.4 | 3.86 (0.96–25.61) | 0.089 |
| GC/aa | 1 | 0.2 | 0 | 0.0 | — | |
| CC/gg | 1 | 0.2 | 3 | 0.6 | 0.32 (0.02–2.52) | 0.326 |
| CC/ga | 0 | 0.0 | 1 | 0.2 | — | |
- a Odds ratios for minor common genotypes or alleles were calculated by comparing each value with the major common genotype or allele.
Parkin Expression Analysis
To verify heterozygous deletions Ex2-3del and Ex5del, real-time PCR quantification of leucocyte Parkin mRNA expression was performed using Ex1-2, Ex4-5, and Ex8-9-specific probes. When the average expression level from seven controls (C1–C7) was set as 1.0, apparent twofold decrease of relative expression level in P1 exon 1–2 region and P2 exon 4–5 region was observed (Fig. 2A–C). To test the relevance of the IVS9 g > a to 1084intron+, leukocyte total and 1084intron+ Parkin mRNA expressions in 15 PD patients were examined using Ex8-9 and intron9-specific probes. The median (range) relative expression value of 1084intron+ mRNA for gg homozygote (n = 8) was lower (but not significantly) than ga heterozygote (n = 6): 0.184 (0.166–0.197) and 0.202 (0.168–0.234), respectively (P = 0.077). The relative expression value for aa homozygote (n = 1) examined was 0.261 (Fig. 2D).

Real-time PCR quantification of leukocyte Parkin mRNA expression. A–C: Expression levels of Parkin Ex1–2, Ex4–5, and Ex8–9 mRNA relative to HPRT1 mRNA. To normalize, the average expression level from seven controls (C1–C7) was set as 1.0. D: Expression levels of Parkin c.1084intron+ RNA level relative to Ex8–9 RNA in leukocytes from IVS9 g > a gg (n = 8), ga (n = 6), and aa (n = 1) genotypes. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
DISCUSSION
Mutation of Parkin is the predominant genetic cause of early onset PD across different ethnic backgrounds. In the present study full-length transcript was RT-PCR amplified using two overlapping fragments and sequenced to reveal changes. Four different heterozygous mutations were identified: one missense change (R334C), two exon deletions (Ex2-3del and Ex5del) and one 86-bp IVS9 insertion (c.1084intron+). The frequency of mutation might be underestimated, since cDNA sequencing would have the potential to miss whole gene deletions or deletions that remove the 5′UTR/transcription start site. Although a loss-of-function mechanism of the Parkin mutations has been indicated and there is some debate over heterozygous mutations as a risk factor for PD, heterozygous mutation carrier status significantly influencing AAO of PD has been reported [Sun et al., 2006]. As mutation screening of PINK1, DJ-1, LRRK2, and ATP13A2 failed to identify a second causative mutation, heterozygosity for a single Parkin mutation may predispose to this disorder in our two patients with deletions. The results of significantly more heterozygous variants in patients with early onset PD than in controls [Lesage et al., 2008] also supports the notion that single Parkin mutations may increase the risk of early onset PD.
The R334C mutation concerns a conserved amino acid located in Parkin IBR (in between RINGs) domain. The mutation causes reduced Parkin solubility and sequestration into an aggresome-like protein aggregates [Wang et al., 2005]. Interestingly, the patient who carries this mutation also bears LRRK2 G2385R and ATP13A2 A746T variants. The three mutations/variants may act together to cause the earlier onset of the clinical symptoms (AAO = 49).
The in-frame Ex2-3del is deduced to remove amino terminal region of 135 amino acids, mainly Ubl domain. The 28 amino acids deleted in Ex5del are located between Ubl and RING1. As E3 activity of Parkin is not affected [Matsuda et al., 2006], the biologic significance of Ex2-3del and Ex5del remains to be determined.
The c.1084intron+ insertion introduces two new amino acids and a TAA stop codon after exon 9 (amino acid residue 361), which may lead to the attenuation of RING2-associated E3 activity [Matsuda et al., 2006] or sequestration into aggresome-like structure [Henn et al., 2005]. The novel c.1084intron+ may result from IVS9 g > a change (Fig. 1B). The expression of 1084intron+ mRNA was higher for aa and ga genotype compared with gg genotype, whereas the correlation of 1084intron+ mRNA expression with IVS9 g > a genotype was not significant (Fig. 2D), which may be due to the small sample size and individual differences. Although how an increased 1084intron+ mRNA expression could modulate risk of PD remains speculative, it is conceivable that the 1084intron+ producing a 361-amino-acid Parkin isoform may result in a dominant-negative like effect and thus increased 1084intron+ expression may be more deleterious.
Two exonic SNPs in the Parkin gene were found and genotyped. No significant difference in 167N allele between patients with PD (38.9%) and controls (40.3%). Our result is consistent with what were found in several previous studies [Wang et al., 1999; Hu et al., 2000; Klein et al., 2000; Oliveri et al., 2001; Lücking et al., 2003; Oliveira et al., 2003b]. For V380L, although several studies reported no association with PD [Wang et al., 1999; Hu et al., 2000; Klein et al., 2000; Oliveri et al., 2001; Oliveira et al., 2003b], there was a trend toward decrease in risk of developing PD for V380L heterozygosity in our study (Table II). This is consistent with the reported significant association of homozygous V380 with sporadic PD [Lücking et al., 2003]. As a functional relationship between Parkin and tau existed [Guerrero et al., 2008], the reported association of 380L with less risk of taupathy progressive supranuclear palsy [Ros et al., 2008] also suggests the functional significance of this variation. Further studies are needed to clarify if and how this polymorphism predisposes to PD.
Acknowledgements
The authors would like to thank the patients and controls for participating in this study. This work was supported by grants NSC-95-2314-B-182A-061 and NSC-96-2314-B-182A-099-MY2 from the National Science Council, Executive Yuan, Taiwan and CMRPG 37056 from Chang Gung Memorial Hospital, Taipei, Taiwan.

