Homozygous microdeletion of the ERI1 and MFHAS1 genes in a patient with intellectual disability, limb abnormalities, and cardiac malformation
Abstract
A male child, born from consanguineous parents and having intellectual disability, short stature, dysmorphic facial features, synpolydactyly, and cardiac malformations is reported. Chromosomal microarray analysis showed that the patient presents with an 8p23.1 homozygous deletion, containing the microRNA miR-4660, the exoribonuclease 1 (ERI1), and malignant fibrous histiocytoma amplified sequence 1 (MFHAS1) genes. The microRNA miR-4660 has no known function. MFHAS1 is an immunomodulatory protein involved in Toll-like receptor signaling, erythropoiesis, and cancer. ERI1 is a ribonuclease involved in RNA metabolism and is required for the correct patterning of the skeleton by defining the HOXC8 expression. We discuss the involvement of these deleted genes to the patient's features and highlight differential diagnoses with syndromes implicating limb extremity abnormalities such as synpolydactyly, including the monosomy 8p.
1 INTRODUCTION
Congenital malformations are single or multiple defects of morphogenesis of organs or body districts, identifiable during the intrauterine life or at birth. Their prevalence is about 2–3% at birth (Corsello & Giuffrè, 2012). Some of the major genetic causes of congenital malformations are copy number variations (CNVs), identified by chromosomal microarray analysis (CMA) (Szczałuba et al., 2016). The latter technique was therefore assigned as a first-tier test in populations of intellectual disability (ID)/developmental delay (DD) or congenital anomalies (Miller et al., 2010).
Here we present a male child, born from consanguineous parents and having ID, dysmorphic facial features, short stature, synpolydactyly, and cardiac malformation. A homozygous microdeletion involving the ERI1 and MFHAS1 genes and the microRNA MIR4660 was found using chromosomal microarray analysis (CMA). Differential diagnoses and possible involvement of the deleted genes in the features presented by the patient are discussed.
2 MATERIALS AND METHODS
2.1 Ethical statement
This study was carried out with protocols approved by the Institutional Review Board (IRB) on human experimentation at the Saint Joseph University, Beirut, Lebanon. Informed written consents were obtained from the patient's parents.
2.2 Clinical report
The male patient here described is the fourth child of healthy consanguineous Lebanese parents. He was born at full term after an uneventful pregnancy, with normal birth length of 50 cm and weight of 3,000 g. He had multiple congenital anomalies, including limb malformations and an atrial septal defect for what he underwent a surgical closure at the age of 5 years.
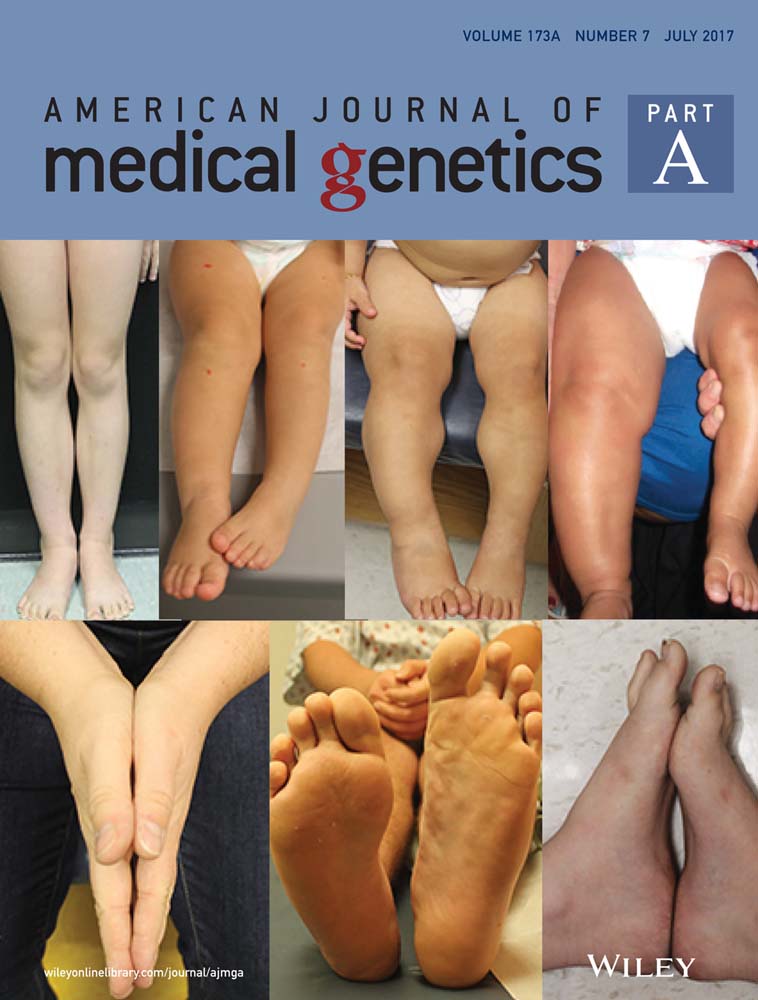
He was referred to us for a clinical examination at the age of 5 years and 6 months. His general cognitive ability was within the below average range of intellectual functioning and his psychomotor and developmental milestones were delayed as the patient sat alone at the age of 9 months and walked without help and started to talk at the age of 28 months. The height was 105 cm (<< 3rd centile) and the head circumference 50.5 cm (10th centile). The dysmorphic facial features consisted of triangular shaped face, flat forehead, high frontal hairline, ptosis of both eyelids, broad nasal root, an arched palate, a pointed chin, and a webbed neck. He had a brachydactyly, a camptodactyly of the 4th right finger, syndactyly with partial webbing of 2/3 and 4/5 left fingers and 3/4 right fingers, and a complete webbing of 2/3, 4/5 left toes, and 4/5 right toes with a partial webbing of 2/3 right toes, as well as nail hypoplasia. X-ray examinations showed short metacarpal bones, short phalanges, abnormally shaped middle phalange bones of all digits giving a close aspect to brachydactyly type A1, and an additional middle metatarsal on both feet with shortening of the left 4th toe (Figure 1). Otherwise, he had a normal skeleton. Abdominal pelvic ultra sound was also normal.

2.3 Chromosomal microarray analysis (CMA)
Chromosomal microarray analysis was performed using the Affymetrix Cytogenetic Whole Genome 2.7M Array. Labeling and hybridization were performed following prescribed Affymetrix ® protocols. The chip was scanned then analyzed using the Affymetrix Chromosome Analysis Suite software (ChAS v.1.0.1). Genomic sequences were studied using the UCSC Genome Browser database (UCSC GRCH36/hg18). The genomic coordinates of the identified microdeletion were afterward converted to correspond to the hg38 build.
2.4 Real-time quantitative PCRs (RQ-PCR)
CNVs were validated by RQ-PCR via the ΔΔCT method, using SYBR Green (Applied Biosystems, Life Technologies, Carlsbad, CA) on an ABI 7500 real time PCR system. RQ-PCRs for the proband, his parents, his healthy brother and sister, and several Lebanese reference samples were run in triplicate. Primers were designed using Primer Express 3 (ABI) for the quantification of ERI1. Adenosine receptor 2b (ADORA2B) was considered as endogenous gene. To exclude the presence of unspecific products, a melting-curve analysis of the products was performed after completion of the amplification.
3 RESULTS
Chromosomal microarray analysis of the patient revealed a homozygous 8p23.1 microdeletion. The minimal size of this deletion, based on the deleted/non-deleted markers on the array, is 284 kb (chr8:8,783,887–9,068,578) (hg38). This region contains two genes ERI1 and MFHAS1 in addition to the microRNA MIR4660 (Figure 2a). No reported regions known to represent nonpathogenic copy number variations and spanning the whole deletion were found in the database of genomic variants (http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg38/).

The homozygous deletion was confirmed in the genomic DNA of the patient using RQ-PCR. The healthy parents, brother, and sister of the patient carried only a single copy of the 8p23.1 deletion (Figure 2b).
4 DISCUSSION
This study provides the first description of an 8p23.1 homozygous microdeletion, found in a patient with ID, psychomotor retardation, dysmorphic facial features, short stature, synpolydactyly, brachydactyly, camptodactyly, nail hypoplasia, and cardiac malformation. The deleted region contains MFHAS1 and ERI1 and the microRNA miR-4660 which has no known function.
The Malignant fibrous histiocytoma-amplified sequence one gene (MFHAS1), also known as (MASL1), belongs to the ROCO proteins family, a multi-domain proteins sharing a conserved ROC-COR supradomain and known to be associated with disorders such as Crohn's disease, cancer or Parkinson disease (Dihanich, 2012). MFHAS1 is an oncoprotein identified from malignant fibrous histiocytomas. Its LRR protein–protein interaction domain points to the MFHAS1 role in signal transduction and control of cell growth in humans (Lewis, 2009; Sakabe et al., 1999). In fact, MFHAS1 regulates erythropoiesis through the Raf/MEK/ERK pathway and has a dual effect (inhibitory and stimulatory), through JNK/NF-κB/AP-1 signaling pathway, on the Toll-like receptor 2 (TLR2) signaling pathway and inflammation (Zhong et al., 2015). Heterozygous deletions of this gene were reported in Decipher patients 288418 and 288554. The first had short stature and short first metacarpals, and the second had a proximal radio-ulnar synostosis (https://decipher.sanger.ac.uk).
The exoribonuclease 1 (ERI1) also known as (THEX1) is involved in fundamental cellular processes such as protein biogenesis, DNA replication and cell division. It has a major impact on three RNA target molecules: ribosomal RNA, histone mRNA, and miRNA (Thomas et al., 2014). ERI1 is highly expressed in mouse bladder, bone, dorsal root ganglion, spleen, and thymus (Rath, 2010; Thomas et al., 2012). It is well conserved from fission yeast to humans (Iida, Kawaguchi, & Nakayama, 2006). Mice homozygous for a null ERI1 allele exhibit postnatal lethality, decreased body size beginning at E15.5, and decreased proliferation of embryonic fibroblasts (Ansel et al., 2008). Further, a skeletal dysmorphology phenotype was found, revealing an additional pair of ribs and a fusion of the eighth rib pair to the sternum in some of these mice. In fact, ERI1 inhibits miR-196-mediated HOXC8 3′UTR silencing in mice (Rath, 2010). HOXC8 is a transcription factor that controls the formation of the primary and secondary body axes during embryonic development. It is known to play a role in the regulation of cartilage differentiation and to be involved in chondrodysplasias or other cartilage disorders (Simeone et al., 1987; Van den Akker et al., 2001; Wan, Shi, Feng, & Cao, 2001). ERI1 defines the HOXC8 expression pattern in the embryo and is consequently required for the correct patterning of the skeleton (Rath, 2010). The short stature and skeletal abnormalities found in the patient described herein could then be related to the complete absence of the MFHAS1 and ERI1 transcripts, the latter possibly leading to an abnormal expression of HOXC8. The study of the expression of HOXC8 in the patient could not be performed as this gene is not expressed in lymphocytes or could be absent in adult cells (http://www.ncbi.nlm.nih.gov/UniGene/EST). No truncation mutations in ERI1 nor in MFHAS1 were reported in ExAC in the homozygous state which supports furthermore the pathogenicity of the deletion.
Our patient's deleted region lies within the critical region of the monosomy 8p23.1 syndrome. Patients having this syndrome present a mild ID, growth deficiency, congenital heart defect, and in some cases, a range of hands and/or feet abnormalities (Baranello et al., 2013; Digilio et al., 1998; Dill, Schertzer, Sandercock, Tischler, & Wood, 1987; Guimiot et al., 2013; Hutchinson, Wilson, & Voullaire, 1992; Rodewald, Stengel-Rutkowski, Schulz, & Cleve, 1977; Scarbrough, Carroll, Finley, & Bridges, 1987; Skrlec, Wagner, Pubeljić, Heffer, & Stipoljev, 2014; Wat et al., 2009; Zafra de la Rosa et al., 2005). The phenotypic resemblance between our patient and probands with the 8p23.1 monosomy, highlights the important role of both ERI1 and MFHAS1 in the limb and cardiac anomalies, and in the developmental delay. Nevertheless, the existence of heterozygous deletions in the healthy consanguineous parents, brother, and sister, and in some 8p monosomy probands, showing no limb nor cardiac malformations, suggests an autosomal recessive mode of inheritance for this feature, which imposes the contribution of additional mutations of MFHAS1 or of the exoribonuclease 1 ERI1 gene or other genes in the occurrence of skeletal malformations, cardiac defects, ID, and developmental delay.
Few syndromes present common features with the patient here described (Table 1). The craniodigital Filippi syndrome (OMIM 272440) is characterized by an intellectual disability, a short stature, heart defects, nail aplasia/dysplasia, syndactyly of the 3rd and 4th fingers and/or the 2nd, 3rd, and 4th toes, a polydactyly, and brachydactyly. These common anomalies to our patient add up to the broad nasal bridge and the high frontal hairline observed (Hussain et al., 2014).
| Main features | Present case | Filippi | Feingold | Tsukahara | Ravel et al. |
|---|---|---|---|---|---|
| OMIM | 272440 | 164280 | 613627 | ||
| Molecular defect | Arr.8p23.1(8,783,887–9,068,578) × 1 | Homozygous or compound heterozygous mutation in CKAP2L | Hemizygous deletion of MIR17HG | ||
| Intellectual disability | + | + | + | + | + |
| Short stature, short limbs | + | + | + | + | + |
| Microcephaly | − | + | + | + | + |
| Hearing impairment | − | − | + | + | + |
| Myopia | − | − | − | + | + |
| Persistent pupillary membrane | − | − | − | + | − |
| Ptosis of eyelids | + | − | − | + | − |
| Bulbous nasal tip | − | − | + | + | + |
| Cleft lip/palate | − | + | + | − | − |
| Scoliosis | − | − | − | + | − |
| Mesomelia of upper limbs | − | − | − | + | − |
| Dislocated elbow | − | − | − | + | − |
| Bowed bones | − | − | − | + | − |
| Brachydactyly | + | + | + | A1 | A4, B, and E |
| Clinodactyly of fingers | − | 5 | 2 and 5 | 2 and 5 | 5 |
| Camptodactyly | +4 right hand | − | − | − | + |
| Fingers syndactyly | 2 and 3 left hand, 3 and 4 right hand | + | − | − | + |
| Dysplastic/short nails | Aplasia of nails: 3 and 5 left hand, 3 and 4 right hand | + | + | + | − |
| Brachytelephalangy | + | − | − | + | + |
| Brachymesophalangy of fingers | − | 5 | 2 and 5 | 2–5 | 2 and 5 |
| Short metacarpals | + | + | − | − | 4 and 5 |
| Fused carpal bones | − | + | + | − | + |
| Toes syndactyly | 2 and 3, 4 and 5 bilateral | + | + | + | + |
| Heart abnormalities | + | + | + | − | + |
| Corpus callosum anomalies | − | − | + | − | + |
| Sacral agenesis | − | − | − | − | + |
In the Feingold syndrome (Feingold, Hall, Lacassie, & Martínez-Frías, 1997) (OMIM 164280) there are abnormal nails and brachymesophalangy of fingers, and heart anomalies however, with different dysmorphic facial features, microcephaly, and a clinodactyly.
Probands having the Tsukahara syndrome (Tsukahara, Azuno, & Kajii, 1989; Utine et al., 2010) are known for ptosis of their eyelids and they show with Ravel et al. (2011) described patient brachytelephalangy and brachydactyly common to our patient. Because of this clinical overlap, it might be interesting to look in patients with such syndromes for mutations in ERI1 and MFHAS1.
In conclusion, we are reporting a patient with ID, short stature, limb, and cardiac malformations, and having a microdeletion of ERI1 and MFHAS1. Additional patients with a similar genotype/phenotype are needed to further investigate the involvement of the deleted genes in the occurrence of cardiac and skeletal defects and most importantly their contribution to the 8p23.1 monosomy syndrome.
ACKNOWLEDGMENTS
We thank the family for their cooperation. This study was supported by the Council of the Saint Joseph University, Beirut, Lebanon.
CONFLICT OF INTEREST
The authors declare no conflict of interest.